03 October 2020: Review Articles
Prospects for Use of Single-Cell Sequencing to Assess DNA Methylation in Asthma
Shuai Men1ABCDEF, Yanyan Yu1ABCEG*DOI: 10.12659/MSM.925514
Med Sci Monit 2020; 26:e925514






Abstract
ABSTRACT: Asthma is a complex disease with an increasing prevalence rate caused by the interaction of multiple genetically inherited and environmental factors. Epigenetics link genetic susceptibility and environmental factors. DNA methylation is an epigenetic modification that plays a crucial role in the regulation of growth and development, gene expression, and disease. Relatively little is known about DNA methylation in asthma, with few studies to date using single-cell sequencing to analyze the molecular mechanism by which DNA methylation regulates asthma. Cells with similar phenotypes may be heterogeneous in function and transcription, as may their genetic information. Although multi-omics methods, such as studies of the genome, transcriptome, and epigenome, can be used to evaluate biological processes, these methods are applicable only to groups of cells or tissues and provide averages that may obscure direct correlations among multiple layers of data. Single-cell sequencing technology can clarify the methylation and expression of genes in different populations of cells, in contrast to traditional multi-omics sequencing, which can determine only average values of cell populations. Single-cell sequence can therefore better reflect the pathogenesis of asthma, as it can clarify the function and regulatory mechanism of DNA methylation in asthma, and detect new genes and molecular markers that may become therapeutic targets in this disease.
Keywords: Asthma, DNA Methylation, Epigenesis, Genetic, Single-Cell Analysis, Epigenomics, Sequence Analysis, DNA
Background
Asthma is a heterogeneous chronic inflammatory airway disease and one of the most common respiratory diseases in children. It is characterized by variable respiratory symptoms, including restricted reversibility of airflow, severe airway remodeling, and even suffocation. In recent years, epidemiological studies in many countries and regions have shown that the prevalence of asthma has increased annually [1], with this disease currently affecting more than 300 million people worldwide.
Asthma is a complex disease influenced by the interaction of genetic (48–79%) and environmental factors [2]. Epigenetic phenomena link environmental and genetic susceptibility factors by influencing gene modification and regulating gene function [3]. DNA methylation, histone modification, and microRNA are the 3 primary mechanisms by which epigenetics are changed by environmental exposure, such as air pollution, diet, drugs, microorganisms, obesity and stress factors. Prenatal exposure to these environmental factors is thought to cause epigenetic changes, thereby affecting early immune programming and influencing the development and function of the immune system, changes that can have lasting effects (Figure 1) [4–9]. Although identifying these factors will be difficult, it is critical to choose the appropriate tissue to study the effect of the epigenome on asthma. Greater cell purity and a higher correlation between cells and disease states increase the likelihood that epigenetic markers will regulate the expression of
The immunological pathogenesis of asthma is closely related to functional imbalances of Th1/Th2 cells and Th2/Treg cells [11] and the hyperfunction of Th2 cells. The differentiation of Th and Treg cells is affected by epigenetic factors, including the characteristics of anti-inflammatory antigen-presenting cells and the structure of antigens. In addition, cytokine gene expression and epigenetics play major roles in regulating asthma airways. Epigenetic mechanisms have been shown to regulate the expression of transcription factors and cytokines during T cell differentiation into Th1, Th2, and Treg cells [12]. Epigenetics can regulate the expression of the Th1 cytokine interferon (IFN)-γ, and reduced IFN-γ expression can inhibit CD4+ T cells in asthma patients. Extensive methylation of interleukin (IL)-4 and IFN-γ promoter regions has been observed in naive T cells of asthmatic patients [13]. In addition, the expression and activity of histone acetyltransferase (HAT) and histone deacetylase (HDAC) are abnormal in asthma patients. Abnormal Notch1 signaling can shift the Th1/Th2 balance toward Th2 in asthma patients. The activity of HAT, an enzyme in the Notch1 pathway, is enhanced, and H3K9, H3K14, H3K27, H3K18, and H3K16 are highly acetylated, resulting in abnormal regulation of Notch1 signals in T cells and aggravated inflammatory reactions [14]. MicroRNAs (miRNAs) can also regulate multiple steps in the pathogenesis of asthma. For example, miRNAs-181a, -150, -146a, and -146b in CD4+ T cells are significantly higher in subjects with than without asthma. MiRNA-146a expression is significantly reduced by dexamethasone treatment; whereas levels of miRNAs-181a, -146a, and -146b were found to correlate positively with the total number of inflammatory cells and eosinophils in bronchoalveolar lavage fluid [15]. Taken together, these findings indicate that epigenetic mechanisms play a crucial part in the pathogenesis of asthma. At present, however, studies on the epigenetics of asthma are still inconclusive, indicating the need for further research to understand epigenetic mechanisms in asthma and to identify new therapeutic targets.
The Role of DNA Methylation in Epigenetics
DNA methylation can affect the onset of various diseases by regulating the body’s immune system. DNA methylation is involved in regulating the differentiation of immune cells and the expression of immune factors, confirming that DNA methylation is involved in regulating the immune system. DNA methylation, the most common type of epigenetic modification, is crucial in regulating genome function [16], including in diseases of the immune system such as asthma. The CpG dinucleotide is an important target of DNA methylation in the human genome. The distribution of CpG dinucleotides in the human genome is uneven, but one of the most notable features is CpG islands, which are located in core sequences of structural gene promoters at sites of initiation of gene transcription in 56% of coding genes [17]. Unmethylated CpG islands allow gene transcription, whereas their methylation directly prevents transcription factors, including AP-2, c-Myc and E2F, from binding to the gene promoter region, reducing or abolishing gene transcription [18]. DNA methylation in mammalian cells is mainly catalyzed by the DNA methyltransferases (DNMTs) DNMT1, DNMT3a, and DNMT3b. Using S-adenosylmethionine as a methyl donor, these enzymes catalyze the covalent binding of an activated methyl group to the C-5 position on the dinucleotide CpG, forming 5-methylcytosine and resulting in methylated CpG (mCpG) (Figure 2) [19]. DNA methylation can also occur at the N-6 position of adenine, the N-4 position of cytosine, or the N-7 position of guanine. Methylation mediates gene silencing through a series of DNA-protein and protein-protein interactions, beginning with the binding of 5-methylcytosine binding domain (MBD) proteins and followed by the recruitment of histone-modifying enzymes, which together promote chromatin condensation and inactivation [19–21]. The MBD family of proteins specifically binds to methylated CpG sites on DNA to repress active transcription. To date, 5 members of this conserved family have been identified, MBD1, MBD2, MBD3, MBD4, and MeCP2 [22]. The binding of MBD proteins to methylated CpG islands in gene promoter regions inhibits the binding of transcription factors and cofactors, allowing HDAC to form a complex with other transcription inhibitors (Sin3a) on lysine residues in the tails of histones, increasing the positive charge on histones. This results in chromosome compaction and changes in conformation, further restricting the binding of transcription factors and leading to the inhibition of gene transcription (Figure 3) [23–26].
DNA Methylation in Asthma
DNA methylation markers can be inherited and/or be affected by environmental factors. Many genes related to methylation in asthma, including those encoding the cytokines IL-4, IL-10, IL-13, and IFN-γ; genes involved in signaling pathways, such as FOXP3, iNOS, and Samd; and other genes, such as RUNX3 and ACSL3 [27–31]. Studies of their methylation patterns can result in better understanding of the regulation of asthma pathogenesis. Moreover, the characteristic changes in the methylation of these key genes are related to exposure to certain risk factors. Environmental exposure in early life can cause changes in DNA methylation levels, altering the regulation of the immune system and leading to the development of asthma and other allergic diseases [32]. Risk factors in the environment mainly affect the occurrence of asthma by acting on the methylation of T cells and dendritic cells (DCs). For example, exposure to
DNA methylation also requires the participation of methyl donors. Vitamins, folic acid, and choline in the diet can provide methyl groups for DNA methylation [38]. The offspring of female rats fed a high-methylated diet during pregnancy were found to have higher levels of allergic airway inflammation, serum IgE, and airway hyperresponsiveness than the offspring of rats fed a low-methylated diet. These studies also showed that the asthma gene is hypermethylated and the transcription levels of related genes are reduced. This immune phenotype, however, is reversed after treatment with demethylating agents [39,40].
Destruction of the normal DNA methylation pattern in a cell alters gene expression. Many studies have verified that DNA methylation plays a crucial part in the pathogenesis of asthma [41–43]. An epigenome association study analyzing the relationship between methylation and asthma found that 14 CpG methylation sites in the blood DNA of asthmatic children were associated with decreased initial T cell activity and increased activity of memory CD8 T cells and natural killer (NK) cells. These CpG sites and transcriptional profiles were associated with the activation of eosinophils and cytotoxic T lymphocytes (CTLs) [44]. Moreover, assessments of peripheral blood mononuclear cells (PBMCs) of asthma patients identified 81 CpG regions with differential methylation status. Some genes related to immunity (IL-13, RUNX3) and T lymphocytes were hypermethylated, and 11 differential methylation sites were associated with high serum IgE concentrations. These hypermethylation sites are involved in the processes of T cell maturation, Th2 cell immunity, and oxygen pressure [31]. Changes in DNA methylation may be crucial to the establishment of asthma-related immune phenotypes. Because the relationships between specific mechanisms and DNA methylation remain unclear, it is particularly important to determine changes in DNA methylation in asthma patients. Most of the current studies of DNA methylation in asthma have assessed specific gene
The Significance of Single-Cell Sequencing
Over the past decade, next-generation sequencing (NGS) technology has provided unprecedented insight into biology and heterogeneity at the single-cell level [45], leading to unprecedented understanding of diseases. These advances in single-cell technology are important because volumetric measurements mask crucial information by averaging signals from cells [46]. In particular, NGS has proven a sensitive monitor of gene expression, epigenetic modifications, chromatin and nuclear structure, and other aspects of cellular status [47], enabling further studies of the genome, transcriptome, epigenome, and proteome at the single-cell level [48–50]. Single-cell sequencing analysis was based on similar techniques developed to analyze bulk cell populations. Because most single-cell sequencing-based analysis requires a minimum level of materials, amplification strategies, and physical capture and isolation of individual cells are particularly important for its development. The amplification and sequencing of genomes or transcriptomes can detect single-nucleotide variants (SNVs) and copy number variants (CNVs), as well as determining single-cell gene expression levels and epigenome DNA methylation states [51], revealing different aspects of the functional state of cells. These methods can be used to study the genetic characteristics of diseases and biological processes at the single-cell level. The ability to measure DNA methylation in single cells may further enable understanding of several crucial biological processes, including disease progression, embryonic development, aging, and stem cell differentiation.
Single-cell epigenetic analysis is a major technical challenge [52,53], as huge differences in epigenetic state are observed even in homogeneous cell populations [54]. Although all cells of a particular type are thought to be identical, evidence from single-cell studies indicates that this assumption is not entirely correct [55,56]. Individual cells of the same type may differ widely in functions and properties, with each cell having different effects on human health. Experiments that evaluate populations of cells may average or dilute differences among seemingly identical cells. Moreover, certain types of cells, such as early embryos, are scarce and cannot be assessed in bulk. As with NGS, the need for a sufficient amount of DNA or RNA is a major obstacle to the direct evaluation of epigenetics in these cells. Therefore, the development of high-throughput methods to detect epigenomes at the single-cell level is of great importance in studying the pathogenesis of diseases. Although single-cell sequencing has been useful in assessments of marine microbial diversity, renal clear cell carcinomas, and bone marrow proliferative tumors [57–59], this technique has not been used in research on allergic diseases such as asthma.
Asthma is a disease related to immune cells. These immune cells have abundant subpopulations, and the type, state, distribution, and function of these cells are polymorphic, allowing them to maintain a steady state of immunity through a complex network of cytokines. Single-cell sequencing can reveal the tiny differences specific to each cell, and exploring the pathogenesis of asthma at the single-cell level can help identify new therapeutic targets.
Application Potential of Single-Cell DNA Methylation Sequencing
The most commonly used techniques for detecting DNA methylation at the single-cell level are single-cell reduced representation bisulfite sequencing (scRRBS) and single-cell bisulfite sequencing (scBS-seq). These methods mainly involve restrictive digestion and bisulfite treatment of CpG-rich regions, allowing the current DNA methylation level of an individual cell to be measured and up to 48% of CpG sites in the genome to be detected [51,53,60–62], a measure that also indirectly reflects the expression of most genes. During the process of single-cell DNA methylation sequencing, it is necessary to separate individual cells. Because of the small amount of nucleic acids in single cells, nucleic acid sequences must be amplified. Single-cell DNA methylation sequencing consists mainly of single-cell isolation, lysis, and bisulfite conversion, followed by library construction, sequencing, and bioinformatics analysis (Figure 4). Cells with similar phenotypes have been shown to be heterogeneous in function and transcription, suggesting that their genetic information may also differ significantly. The DNA used in traditional high-throughput sequencing is extracted from populations of cells, with the results being only the average of this cell population. This overall characterization approach masks much low-abundance information [63,64]. Because single-cell sequencing technology can more truly reflect the occurrence of disease and its mechanism of development, single-cell DNA methylation is increasingly used in research on diseases. Single-cell whole-genome methylation technology has been used to study increases in CpG methylation during the maturation of human oocytes, which may enable assessments of DNA methylation reprogramming in early human embryos [65,66]. Analysis of the heterogeneity of 5-methylcytosine patterns in the whole liver of mice by assessing single-cell DNA methylomes showed a significantly high level of heterogeneity, corresponding to an average mutation frequency of about 3.3%, The region containing H3K4me1 was found to be the most variable, the promoter and CpG islands to be the most stable, most non-functional sites such as repeating elements and introns to be unstable, and the potential functional sites such as gene promoters to be generally stable [67]. Single-cell technology has been used to assess hematopoietic stem cell subpopulations [68], splicing variability and cell heterogeneity [69], and epigenetic chimerism in preimplantation embryos [70]. Findings to date suggest that this technology will be useful in assessing the pathogenesis of asthma, rhinitis, and other diseases and in providing a basic understanding of complex biological phenomena.
Although this technology is still in the laboratory research stage, we think it may have good clinical application. For example, it can be used to analyze the same cells of different organs or different types of cells in the same organ, such as differences in methylation of blood lymphocytes and in the alveolar lavage fluid of patients with asthma; to detect differences in the DNA methylation status of various types of cells in asthma patients; and to detect immune cell heterogeneity in asthma patients. This method may also identify new subtypes of immune cells, enhancing individualized treatment.
Conclusions
Epigenetics are reversible genetic changes that can silence or activate the expression of specific genes through various modifications. It is an important link between the environment and genetics. Because epigenetics play a role in immune system development and regulation of function, epigenetics, especially DNA methylation, have an important impact on the occurrence of asthma. Few studies to date have utilized single-cell sequencing to assess the regulation of DNA methylation in diseases, especially the mechanism by which DNA methylation regulates asthma. Single-cell sequencing not only provides a more comprehensive understanding of the regulatory role of DNA methylation in asthma, including gene expression and immune cell differentiation, but can help identify critical surface markers in asthma-related immune cells. These genes and molecular markers may become a new target for immunotherapy and have important value in clinical research.
Figures
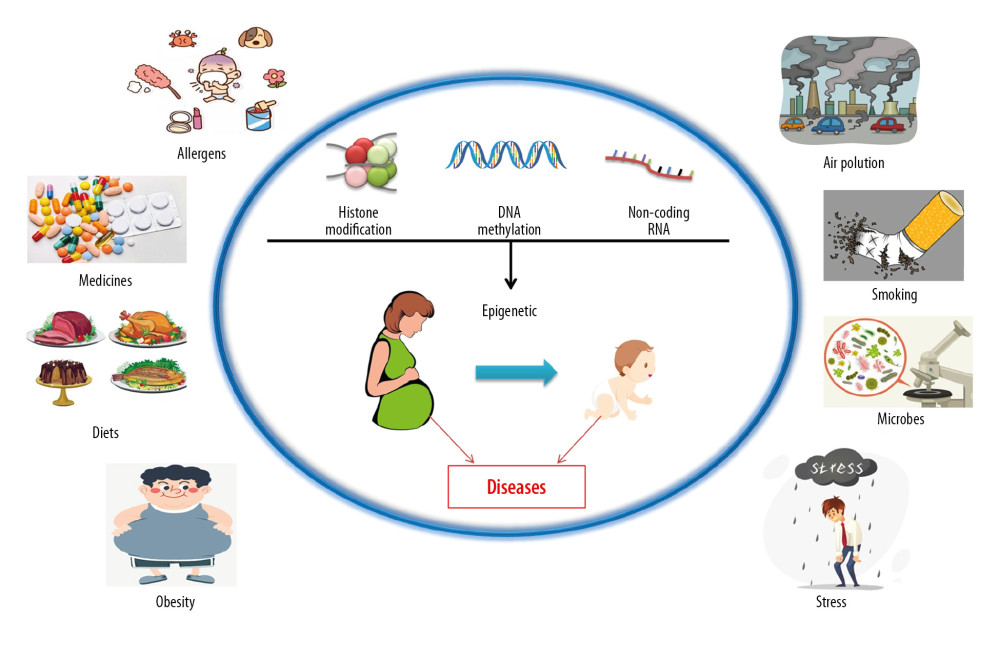 Figure 1. Epigenetic modifications by which environmental exposure influences the occurrence of diseases.
Figure 1. Epigenetic modifications by which environmental exposure influences the occurrence of diseases. 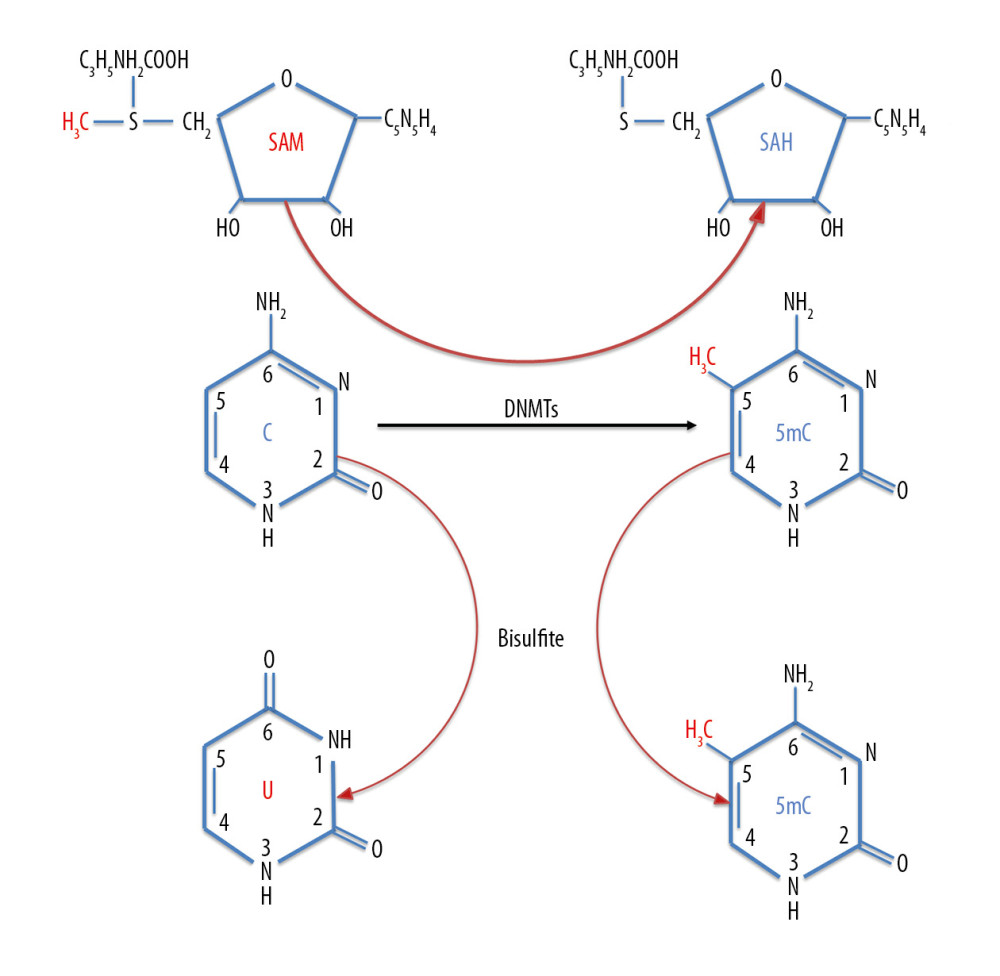 Figure 2. DNA methylation process.
Figure 2. DNA methylation process. 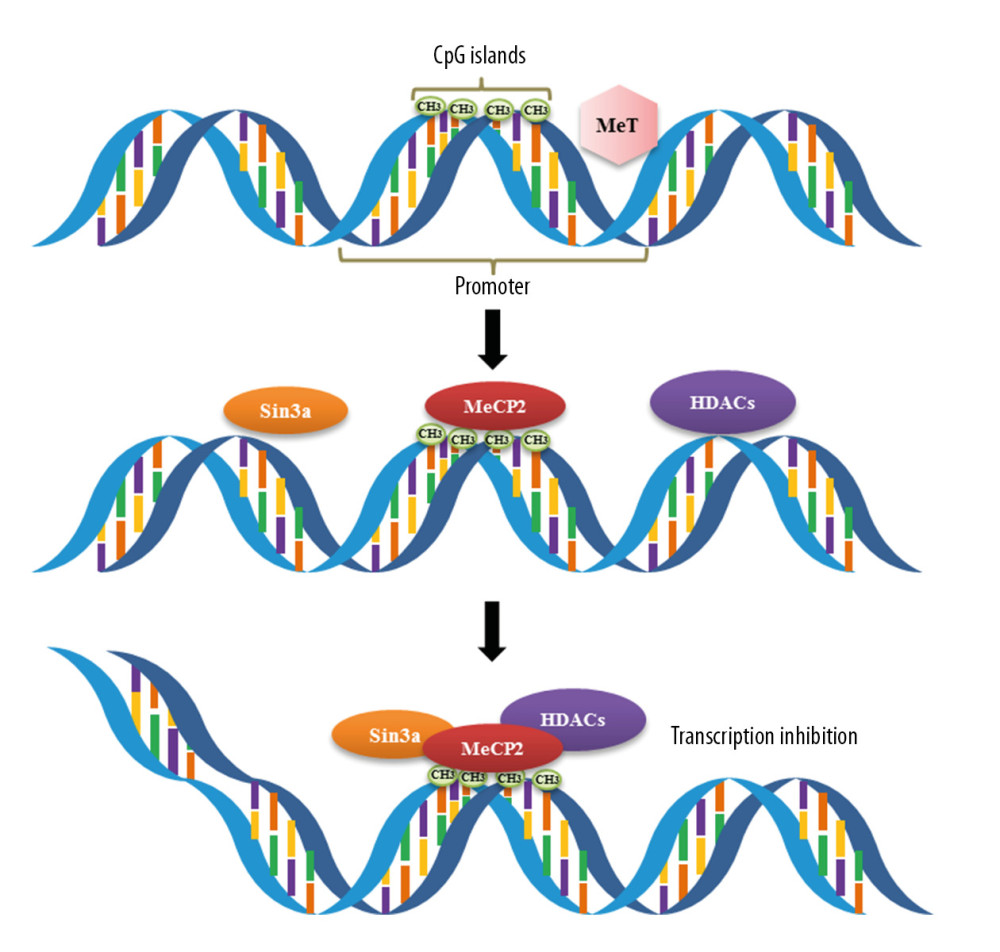 Figure 3. The main mechanism by which DNA methylation inhibits gene transcription.
Figure 3. The main mechanism by which DNA methylation inhibits gene transcription. 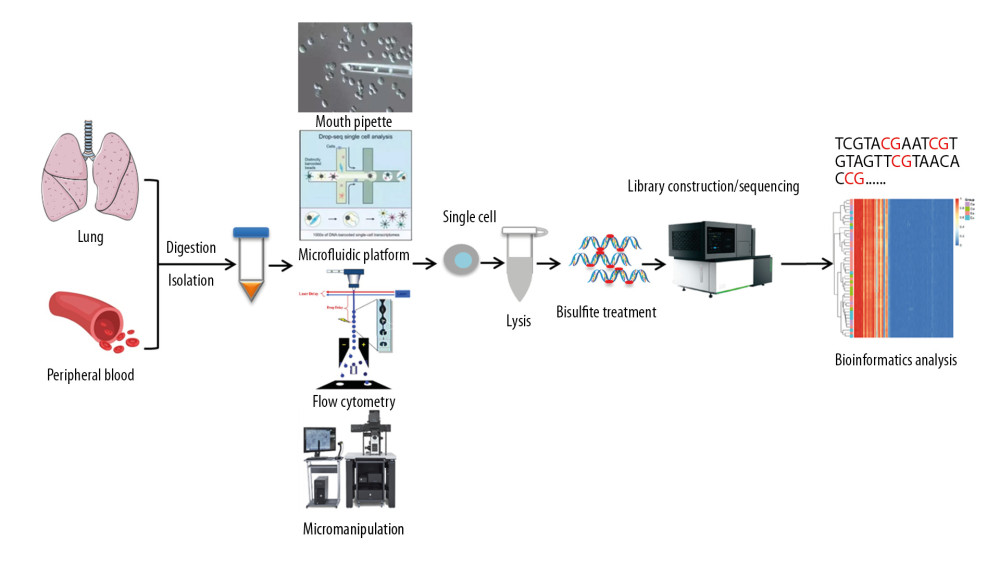 Figure 4. Single-cell DNA methylation sequencing technology in asthma.
Figure 4. Single-cell DNA methylation sequencing technology in asthma. References
1. Ellwood P, Asher MI, Billo NE, The Global Asthma Network rationale and methods for Phase I global surveillance: Prevalence, severity, management and risk factors: Eur Respir J, 2017; 49(1); 1601605
2. Pinto LA, Stein RT, Kabesch M, Impact of genetics in childhood asthma: J Pediatr (Rio J), 2008; 84(4 Suppl); S68-75
3. Salam MT, Asthma epigenetics: Adv Exp Med Biol, 2014; 795; 183-99
4. Sabounchi S, Bollyky J, Nadeau K, Review of environmental impact on the epigenetic regulation of atopic diseases: Curr Allergy Asthma Rep, 2015; 15(6); 33
5. Burte E, Nadif R, Jacquemin B, Susceptibility factors relevant for the association between long-term air pollution exposure and incident asthma: Curr Environ Health Rep, 2016; 3(1); 23-39
6. Viljoen K, Segurado R, O’Brien JDMed on behalf of the Lifeways Cross Generation Cohort Study Steering Group, Pregnancy diet and offspring asthma risk over a 10-year period: The Lifeways Cross Generation Cohort Study, Ireland: BMJ Open, 2018; 8(2); e017013
7. Ege MJ, Mayer M, Normand AC, Exposure to environmental microorganisms and childhood asthma: N Engl J Med, 2011; 364(8); 701-9
8. Peters U, Dixon AE, Forno E, Obesity and asthma: J Allergy Clin Immunol, 2018; 141(4); 1169-79
9. Flanigan C, Sheikh A, Dunngalvin A, Prenatal maternal psychosocial stress and offspring’s asthma and allergic disease: A systematic review and meta-analysis: Clin Exp Allergy, 2018; 48(4); 403-14
10. Yang IV, Schwartz DA, Epigenetic mechanisms and the development of asthma: J Allergy Clin Immunol, 2012; 130(6); 1243-55
11. Yang YL, Pan YQ, He BS, Zhong TY, Regulatory T cells and Th1/Th2 in peripheral blood and their roles in asthmatic children: Transl Pediatr, 2013; 2(1); 27-33
12. Yang IV, Lozupone CA, Schwartz DA, The environment, epigenome, and asthma: J Allergy Clin Immunol, 2017; 140(1); 14-23
13. Kwon NH, Kim JS, Lee JY, DNA methylation and the expression of IL-4 and IFN-gamma promoter genes in patients with bronchial asthma: J Clin Immunol, 2008; 28(2); 139-46
14. Cui ZL, Gu W, Ding T: Int J Immunopathol Pharmacol, 2013; 26(2); 371-81
15. Feng MJ, Shi F, Qiu C, Peng WK: Int Immunopharmacol, 2012; 13(3); 347-53
16. Schübeler D, Function and information content of DNA methylation: Nature, 2015; 517(7534); 321-26
17. Gunawardhana LP, Gibson PG, Simpson JL, Characteristic DNA methylation profiles in peripheral blood monocytes are associated with inflammatory phenotypes of asthma: Epigenetics, 2014; 9(9); 1302-16
18. Yin Y, Morgunova E, Jolma A, Impact of cytosine methylation on DNA binding specificities of human transcription factors: Science, 2017; 356(6337); eaaj2239
19. Ginder GD, Williams DC, Readers of DNA methylation, the MBD family as potential therapeutic targets: Pharmacol Ther, 2018; 184; 98-111
20. Shimbo T, Wade PA, Proteins that read DNA methylation: Adv Exp Med Biol, 2016; 945; 303-20
21. Valinluck V, Tsai HH, Rogstad DK, Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2): Nucleic Acids Res, 2004; 32(14); 4100-8
22. Liu K, Xu C, Lei M, Structural basis for the ability of MBD domains to bind methyl-CG and TG sites in DNA: J Biol Chem, 2018; 293(19); 7344-54
23. Jones PA, Takai D, The role of DNA methylation in mammalian epigenetics: Science, 2001; 293(5532); 1068-70
24. Klose RJ, Bird AP, MeCP2 behaves as an elongated monomer that does not stably associate with the Sin3a chromatin remodeling complex: J Biol Chem, 2004; 279(45); 46490-96
25. Nan X, Ng HH, Johnson CA, Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex: Nature, 1998; 393(6683); 386-89
26. Nikitina T, Shi X, Ghosh RP, Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin: Mol Cell Biol, 2007; 27(3); 864-77
27. Christensen S, Jaffar Z, Cole E, Prenatal environmental tobacco smoke exposure increases allergic asthma risk with methylation changes in mice: Environ Mol Mutagen, 2017; 58(6); 423-33
28. Perera F, Tang WY, Herbstman J, Relation of DNA methylation of 5′-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma: PLoS One, 2009; 4(2); e4488
29. Zhu X, Chen Q, Liu Z, Low expression and hypermethylation of FOXP3 in regulatory T cells are associated with asthma in children: Exp Ther Med, 2020; 19(3); 2045-52
30. Torrone DZ, Kuriakose JS, Moors K, Reproducibility and intraindividual variation over days in buccal cell DNA methylation of two asthma genes, interferon γ (IFNγ) and inducible nitric oxide synthase (iNOS): Clin Epigenetics, 2012; 4(1); 3
31. Yang IV, Pedersen BS, Liu A, DNA methylation and childhood asthma in the inner city: J Allergy Clin Immunol, 2015; 136(1); 69-80
32. Tanday S, Epigenetic study identifies genes linked to asthma and allergy: Lancet Respir Med, 2015; 3(4); 274
33. Vercelli D, Genetics, epigenetics, and the environment: Switching, buffering, releasing: J Allergy Clin Immunol, 2004; 113(3); 381-87
34. Liu J, Ballaney M, Al-alem U, Quan C: Toxicol Sci, 2008; 102(1); 76-81
35. Nadeau K, McDonald-Hyman C, Noth EM, Ambient air pollution impairs regulatory T-cell function in asthma: J Allergy Clin Immunol, 2010; 126(4); 845-52
36. Kim HP, Leonard WJ, CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: A role for DNA methylation: J Exp Med, 2007; 204(7); 1543-51
37. Wen H, Schaller MA, Dou Y, Dendritic cells at the interface of innate and acquired immunity: The role for epigenetic changes: J Leukoc Biol, 2008; 83(3); 439-46
38. Jia M, Gao X, Zhang Y, Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: A systematic review: Clin Epigenetics, 2016; 8; 25
39. Devries A, Vercelli D, Epigenetics of human asthma and allergy: Promises to keep: Asian Pac J Allergy Immunol, 2013; 31(3); 183-89
40. Han YY, Blatter J, Brehm JM, Diet and asthma: Vitamins and methyl donors: Lancet Respir Med, 2013; 1(10); 813-22
41. Breton CV, Byun HM, Wang X, DNA methylation in the arginase-nitric oxide synthase pathway is associated with exhaled nitric oxide in children with asthma: Am J Respir Crit Care Med, 2011; 184(2); 191-97
42. Baccarelli A, Rusconi F, Bollati V, Nasal cell DNA methylation, inflammation, lung function and wheezing in children with asthma: Epigenomics, 2011; 4(1); 91-100
43. Reese SE, Xu CJ, den Dekker HT, Epigenome-wide meta-analysis of DNA methylation and childhood asthma: J Allergy Clin Immunol, 2019; 43(6); 2062-74
44. Xu CJ, Söderhäll C, Bustamante M, DNA methylation in childhood asthma: An epigenome-wide meta-analysis: Lancet Respir Med, 2018; 6(5); 379-88
45. Trapnell C, Defining cell types and states with single-cell genomics: Genome Res, 2015; 25(10); 1491-98
46. Philpott M, Cribbs AP, Brown T, Advances and challenges in epigenomic single-cell sequencing applications: Curr Opin Chem Biol, 2020; 57; 17-26
47. Stadhouders R, Filion GJ, Graf T, Transcription factors and 3D genome conformation in cell-fate decisions: Nature, 2019; 569(7756); 345-54
48. Buenrostro JD, Wu B, Litzenburger UM, Single-cell chromatin accessibility reveals principles of regulatory variation: Nature, 2015; 523(7561); 486-90
49. Palii CG, Cheng Q, Gillespie MA, Single-cell proteomics reveal that quantitative changes in co-expressed lineage-specific transcription factors determine cell fate: Cell Stem Cell, 2019; 24(5); 812-20
50. Buenrostro JD, Corces MR, Lareau CA, Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation: Cell, 2018; 173(6); 1535-48
51. Liang J, Cai W, Sun Z, Single-cell sequencing technologies: current and future: J Genet Genomics, 2014; 41(10); 513-28
52. Schwartzman O, Tanay A, Single-cell epigenomics: Techniques and emerging applications: Nat Rev Genet, 2015; 16(12); 716-26
53. Clark SJ, Lee HJ, Smallwood SA, Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity: Genome Biol, 2016; 17; 72
54. Kelsey G, Stegle O, Reik W, Single-cell epigenomics: Recording the past and predicting the future: Science, 2017; 358(6359); 69-75
55. Cai L, Friedman N, Xie XS, Stochastic protein expression in individual cells at the single molecule level: Nature, 2006; 440(7082); 358-62
56. Rosenfeld N, Young JW, Alon U, Gene regulation at the single-cell level: Science, 2005; 307(5717); 1962-65
57. Yoon HS, Price DC, Stepanauskas R, Single-cell genomics reveals organismal interactions in uncultivated marine protists: Science, 2011; 332(6030); 714-17
58. Xu X, Hou Y, Yin X, Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor: Cell, 2012; 148(5); 886-95
59. Hou Y, Song L, Zhu P, Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm: Cell, 2012; 148(5); 873-85
60. Farlik M, Sheffield NC, Nuzzo A, Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics: Cell Rep, 2015; 10(8); 1386-97
61. Guo H, Zhu P, Guo F, Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing: Nat Protoc, 2015; 10(5); 645-59
62. Smallwood SA, Lee HJ, Angermueller C, Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity: Nat Methods, 2014; 11(8); 817-20
63. Navin N, Hicks J, Future medical applications of single-cell sequencing in cancer: Genome Med, 2011; 3(5); 31
64. Macaulay IC, Voet T, Single cell genomics: Advances and future perspectives: PLoS Genet, 2014; 10(1); e1004126
65. Yu B, Dong X, Gravina S, Genome-wide, single-cell DNA methylomics reveals increased non-CpG methylation during human oocyte maturation: Stem Cell Rep, 2017; 9(1); 397-407
66. Zhu P, Guo H, Ren Y, Single-cell DNA methylome sequencing of human preimplantation embryos: Nat Genet, 2018; 50(1); 12-19
67. Gravina S, Dong X, Yu B, Vijg J, Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome: Genome Biol, 2016; 17(1); 150
68. Hui T, Cao Q, Wegrzyn-Woltosz J, High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations: Stem Cell Rep, 2018; 11(2); 578-92
69. Linker SM, Urban L, Clark SJ, Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity: Genome Biol, 2019; 20(1); 30
70. Lorthongpanich C, Cheow LF, Balu S, Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos: Science, 2013; 341(6150); 1110-12
In Press
08 Mar 2024 : Clinical Research
Evaluation of Foot Structure in Preschool Children Based on Body MassMed Sci Monit In Press; DOI: 10.12659/MSM.943765
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952