14 December 2020: Review Articles
Cell Sources and Influencing Factors of Liver Regeneration: A Review
Chengzhan Zhu12AEG, Bingzi Dong2FG, Leqi Sun3EF, Yixiu Wang1BCE, Shuhai Chen4AEF*DOI: 10.12659/MSM.929129
Med Sci Monit 2020; 26:e929129






Abstract
ABSTRACT: Liver regeneration (LR) is a set of complicated mechanisms between cells and molecules in which the processes of initiation, maintenance, and termination of liver repair are regulated. Although LR has been studied extensively, there are still numerous challenges in gaining its full understanding. Cells for LR have a wide range of sources and the feature of plasticity, and regeneration patterns are not the same under different conditions. Many patients undergoing partial hepatectomy develop cirrhosis or steatosis. The changes of LR in these cases are not clear. Many types of cells participate in LR. Hepatocytes, biliary epithelial cells, hepatic progenitor cells, and human liver stem cells can serve as the cell sources for LR. However, different types and degrees of damage trigger the response from the most suitable cells. Exploring the cell sources of LR is of great significance for accelerating recovery of liver function under different pathological patterns and developing a cell therapy strategy to cope with the shortage of donors for liver transplantation. In clinical practice, the background of the liver influences regeneration. Fibrosis and steatosis create different LR microenvironments and signal molecule interaction patterns. In addition, factors such as partial hepatectomy, aging, platelets, nerves, hormones, bile acids, and gut microbiota are widely involved in this process. Understanding the influencing factors of LR has practical value for individualized treatment of patients with liver diseases. In this review, we have summarized recent studies and proposed our views. We discuss cell sources and the influential factors on LR to help in solving clinical problems.
Keywords: Aging, Fatty Liver, Hepatectomy, Liver Regeneration, Hepatocytes, stem cell niche, Stem Cells
Background
Liver regeneration (LR) involves complicated mechanisms and a tremendous amount of detail. Even after more than 100 years of extensive studies, the process of LR is not completely understood. Currently, the main points of LR include the following: (1) LR is a complicated process involving numerous intrahepatic and extrahepatic components and a large number of signal molecules in a specific microenvironment. Owing to the extremely important functions of the liver, the interaction modules constituted by the saturated signaling pathways rigidly regulate the initiation, maintenance, and termination of LR to achieve homeostasis even in cases of severe injury [1]. (2) Surprisingly, the human liver can regrow with no or almost no loss of function and can survive after sustaining huge damage. This is because of the extensive cell sources and the cell plasticity of LR. According to the level of liver injury and different intrahepatic backgrounds, such as liver fibrosis and liver steatosis, LR can occur in different patterns [2]. (3) The human body is a complicated organism that participates in all life activities as a whole. Various extrahepatic factors, such as partial hepatectomy (PH), aging, platelets, nerves, hormones, bile acid (BA), and gut microbiota can affect LR, thereby creating difficulties in basic research.
These areas of research in LR are challenges, as well as hotspots and key areas of breakthrough research in the field of LR. Therefore, we have summarized the latest advancements in LR research and proposed our interpretations.
Cell Sources for LR
PLASTICITY OF HEPATOCYTES IN HEPATOCYTE-MEDIATED LR:
Different from epithelial tissues such as those in the intestinal tract and skin, which have high proliferative capacity, mature hepatocytes can re-enter the cell cycle for mitosis to complete self-renewal or maintain liver homeostasis after injury without the involvement of progenitor cells [3]. Wang et al. reported that Axin2+ hepatocytes located around the central vein had stem cell-like characteristics [4]. Pu et al. discovered that the Mfsd2a+ hepatocytes in the portal vein triad region are low in number at the hepatic homeostasis phase. However, they could apparently be activated upon PH or chronic liver injury and could extend toward the central area of the liver lobule to replace the injured cells, where their metabolic features were re-edited by the regional microenvironment [5]. Recently, Schaub et al. discovered that a portion of hepatocytes surrounding the portal vein could directly differentiate into bile duct cells, a process that is related to Notch and transforming growth factor-β (TGF-β) signaling [6]. By investigating the sex-determining region Y-box 9 (Sox9) in green fluorescent protein (GFP) transgene-labeled mice, Li et al. found that a hybrid type of hepatocytes are located near the branches of the portal vein and terminal biliary tract, which have the mixed phenotypes of both the hepatocytes and the biliary epithelial cells (BECs) with positive Sox9 and hepatocyte nuclear factor 4α (HNF4α) expression. These hybrid hepatocytes can become hepatocytes and BECs after liver injury to refill the damaged hepatic lobules without increasing the morbidity of cancer [7,8]. Ang et al. found that Lgr5 expression occurs only in a unique subset of hepatocytes that are most adjacent to the central veins. These pericentral Lgr5+ hepatocytes have a long life and contribute to the homeostasis of the liver. After PH, they could fuel the regeneration of their own lineage during massive and rapid LR [9]. Lin et al. found that hepatocytes marked by telomerase expression are sparsely distributed in different locations of the liver lobules. These cells have been shown to be the main source of hepatocytes in normal liver homeostasis and injury-induced LR [10]. These findings demonstrate that the mature hepatocytes have the characteristics of high replication capacity and plasticity (summarized in Figure 1).
MOLECULES AND SIGNALS IN HEPATOCYTE-MEDIATED LR:
In the hepatocyte-mediated LR process, BECs, Kupffer cells, hepatic stellate cells (HSCs), liver sinusoidal endothelial cells, and extrahepatic cells interact. Additionally, blood flow stress signals, immune factors, nerves, hormones, BA, and microbiota are involved (summarized in Figure 2). Under the regulation of various pro-proliferation factors and anti-proliferation factors, the liver initiates and terminates regeneration in a timely way, thus achieving what is called the “hepatostat” [11]. In the classic LR model induced by PH, increased blood flow activates the urokinase plasminogen activator and matrix metalloproteinase to induce the degradation of the extracellular matrix (ECM). Then, the hepatocyte growth factors (HGF) are released from the ECM. Lipopolysaccharides produced by inflammatory response bind with TLR4 receptors on Kupffer cells to release tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6). HSCs and liver sinusoidal endothelial cells can also produce new HGF. Together with the growth factors and cytokines such as the epidermal growth factor (EGF) brought by the portal vein flow, liver mitogens reach a high concentration in the designated site [12]. HGF binds with its receptor C-Met, while TGF-α and EGF bind with their common receptor, EGF receptor (EGFR), to initiate multiple transduction pathways in the hepatocytes, including Wnt/β-catenin, STAT3, and NF-κB [13]. IL-6 can form an excited complex with soluble IL-6R and gp130, which can thereby activate the JAK/STAT, MAPK, and PI3K/AKT pathway in the hepatocytes and regulate the apoptosis-inhibiting nitric oxide synthase to regulate LR [14]. The above pro-proliferation substances induce the sequential formation of complexes between cyclin-dependent kinase (CDK) in the hepatocyte nucleus and cyclins (including cyclin D1 and cyclin E) to mediate the initiation and maintenance of LR [13]. Additionally, angiogenesis is an important step in LR. Angiogenin-2 is downregulated in the initial stage of LR but is upregulated at the angiogenesis phase, thereby regulating the vascular endothelial growth factor to promote angiogenesis. Data have also shown liver sinusoidal endothelial cells could regulate the proliferation of hepatocytes through controlling the synthesis of angiopoietin-2 [15].
When the volume of the regenerated liver reaches the predetermined proportion, the regulatory mechanism of regeneration termination is activated. TGF-β, the most significant hepatocyte proliferation inhibitor and termination signal during the LR process, can regulate DNA synthesis and cell proliferation through the SMAD signaling pathway and initiate the termination of LR. Although TGF-β is upregulated in the early stage of LR, the reduced TGF-β receptor suppresses the production of its anti-proliferation effect [16]. Additionally, high IL-6 expression can rapidly upregulate SOCS3. However, SOCS3 can block STAT3 phosphorylation to inhibit the IL-6 signaling pathway [17]. Such a negative feedback loop demonstrates the homeostasis of LR. The Hippo kinase signal cascade pathway can regulate the YAP/TAZ transcription factors through the MST1/2-LATS1/2 kinases, thereby participating in terminating the LR [18]. Modifying the targets in the Hippo pathway can lead to excessive growth of hepatocytes and cancer predisposition [19]. YAP can also drive the activation of HSCs and substrate synthesis, revealing that the Hippo signal is involved in regulating the homeostasis of LR and liver cirrhosis [20]. Other important LR mediators that have been extensively discussed recently are summarized in the Table 1 [21–38].
Numerous signal transduction pathways in the LR are interdependent and collaborative to make sure that the inactivation of a single pathway will not cause the complete failure of LR. However, this mechanism is not unlimited. Paranjpe et al. discovered that LR was completely inhibited when C-Met and EGFR were blocked [39]. Additionally, during the termination step, the blocking of the TGF-βII receptor in the hepatocytes combined with the inhibition of activin A or the activin A receptor caused the persistent proliferation of hepatocytes [40].
BILIARY EPITHELIAL CELL-MEDIATED LR:
During the embryogenesis process, hepatocytes and BECs are commonly derived from the differentiation of hepatoblasts. De Jong et al. [41] reported that the cells of the peribiliary gland prevent biliary tract loss in the extrahepatic bile duct through proliferation, differentiation, and maturation. Deng et al. [42] utilized the pedigree tracing experiment of the dual-luciferase reporter system and determined that BECs markedly promoted hepatocyte regeneration in 2 chronic liver injury mouse models, and the bile duct cells directly transform into hepatocytes without any intermediate progenitor. Raven et al. discovered that inhibition of hepatocyte proliferation through integrin beta 1 knockdown or p21 overexpression induces the occurrence of bile duct cell-derived hepatocytes [43]. Choi et al. observed in a zebrafish model that the selective pressure caused by excessive consumption of hepatocytes significantly promotes the conversion of BECs into hepatocytes [44]. These findings show that mature BECs could directly differentiate into hepatocytes without transforming into progenitor cells. As a result, we believed that mature BECs could also have high replication capacity and plasticity. Either BECs or hepatocytes are supposed to serve as the cell source for the other when the independent proliferation process is limited.
HEPATIC PROGENITOR CELL-MEDIATED LR:
In 1957, Popper et al. [45] first described the ductular reaction, which manifests as the small bile duct activation and reactive dilatation hyperplasia induced by liver injury. It has been widely demonstrated that hepatic progenitor cells (HPCs) (also referred to as liver progenitor cells, liver stem progenitor cells, and hepatic stem progenitor cells) are the major cells involved in ductular reaction. HPCs can serve as the hepatocyte reservoir in the human liver to achieve an alternative pattern of LR when the canonical pattern of LR is restricted or has failed due to severe liver injury. HPCs, which are called hepatic oval cells in rodents, are the bipotent progenitor cells residing in the root of the most terminal Hering duct of the intrahepatic bile duct. Under the stimulation of injury, quiescent HPCs can proliferate and move from the surrounding hepatic lobules into the hepatic cords, differentiate into 2 major epithelial cells (hepatocytes and BECs), and, finally, fuse to reconstruct the hepatic lobules near the central vein. However, owing to the heterogeneity of HPC-markers in different damage patterns and species, the origin of HPCs remains unclear. HPCs express biliary tract marker cytokeratin (CK) 7/19, but the expression of hepatocyte markers HepPar-1 and HNF4α is also frequently observed [46–48]. When massive necrosis of the liver occurs, the surviving hepatocytes can temporarily de-differentiate into the hepatocytes that express the α-fetoprotein and possess their proliferative features [49]. In a chronically injured liver, mature hepatocytes experience irreversible ductal metaplasia and undergo regeneration to replenish as many as 60% of lost cells [50]. He et al. also verified in a zebrafish model with an almost complete loss of hepatocytes that BECs finally transdifferentiated into hepatocytes through a step of dedifferentiation into a bipotential intermediate [51]. Lu et al. found that in the case of most hepatocytes undergoing aging or apoptosis, the HPCs of biliary origin contributed to completing the functional hepatic reconstruction in mice [52]. The above evidence suggests that the HPCs may not constitute only resident undifferentiated cell clusters. In addition, the mature hepatocytes or bile duct cells could undergo transformation into the opposite lineage, and the intermediates during such a trans-differentiation process may also be sources of the HPCs or HPC-like cells (summarized in Figure 3). Additionally, HPCs can be regarded as the dynamic stem cells that are able to express different markers depending on the lack of epithelial cell type and different pathological features of injury, thus differentiating into different cells. For instance, they undergo differentiation into hepatocytes in the case of severe loss of hepatocytes, such as in acute and chronic viral hepatitis, alcoholic hepatitis cirrhosis, non-alcoholic steatohepatitis, and extensive hepatectomy [46]. By contrast, HPCs become bile duct cells when cholestasis occurs, as during primary biliary cirrhosis and primary sclerotic cholangitis [53]. The concept of the “HPC niche” has been increasingly acknowledged and is used to describe the microenvironment containing extensive interactions between parenchymal and non-parenchymal components during the activation, proliferation, and differentiation of HPCs. Damaged hepatocytes can directly activate HPCs; macrophages, mononuclear cells, and T lymphocytes secrete the (TNF)-like weak inducer of apoptosis; macrophages secrete IL-6 and TNF-α; HSCs secrete matrix metalloproteinase, fibroblast growth factor (FGF), HGF, TGF-α, TGF-β, and TNF-β; liver sinusoidal endothelial cells secrete SDF-1 and vascular endothelial growth factor; and cytokines chelated in the ECM are released by urokinase plasminogen activator and matrix metalloproteinases. All these factors play crucial roles in the activation and amplification of HPCs [54,55]. HGF secreted by the HSCs can bind with MET to activate the PI3K-AKT-STAT3 pathway; oncostatin M secreted by macrophages can activate STAT3; and Wnt3/β-catenin signal is activated by macrophages. These signals control the differentiation of HPC to hepatocytes via the intermediate hepatocytes [54–56]. The Notch signal plays a key role in BEC differentiation. Under the mediation of Jagged1, the activated Notch1 signaling pathway precisely controls the proliferation and differentiation fate of HPCs into BECs via the responsive small bile ducts. Meanwhile, suppression of the Notch pathway can promote the differentiation of HPCs into hepatocytes [56,57] (summarized in Figure 3).
HUMAN LIVER STEM CELL-MEDIATED LR:
Human liver stem cells (HLSCs) were first isolated from fetal liver [58], and then several researches reported that they had successfully isolated HLSCs from adult livers [59–63]. Due to the lack of a unified name, the terms human liver stem cells (HLSCs), adult-derived human liver stem/progenitor cells, and hepatic mesenchymal stem cells have referred to this cluster with self-renewal ability and multilineage differentiation potential. HLSCs express classic stem cell markers, including OCT4, NANOG, SOX2, Vimentin, and Nestin, and typical mesenchymal stem cell markers, including CD29, CD73, CD44, CD90, CD105, and CD166. But they are negative for the hematopoietic stem cell markers CD34, CD45, CD117 and cytokeratin-19, indicating that they are not from the same source as hepatic progenitor cells [59,60,64]. In addition, HLSCs are different from mesenchymal stem cells, especially in terms of their markers and biological functions [65]. HLSCs express markers of immature hepatocytes, including CD26, albumin, α-fetoprotein, CK-8, and CK-18, and present a better hepatocyte differentiation ability and lower immunogenicity than mesenchymal stem cells [59,60,62,66–68]. HLSCs have been confirmed to facilitate LR in several ways. They can be effectively induced into hepatocytes in vitro, or spontaneously differentiated into mature hepatocytes in the damaged liver to promote LR in a liver failure animal model [59,69,70]. Moreover, intraperitoneal injection of conditioned medium obtained from HLSCs significantly increased the proliferation of hepatocytes and upregulated the pro-angiogenesis factor gene in mice that underwent a 70% hepatectomy [71]. The abilities of HLSCs to regenerate a damaged liver can also be achieved through antiinflammatory activity, immune regulation, and anti-fibrosis therapy [65]. At present, the use of HLSCs to treat pediatric patients with metabolic abnormalities has been clinically adopted. Owing to their varied and profound functions on LR, HLSCs are worthy of further exploration [66].
Factors Influencing LR
FIBROSIS:
A variety of liver parenchymal diseases such as viral, alcoholic, and autoimmune hepatitis can induce fibrosis change. Dubuquoy et al. verified [72] that the efficiency of HPCs to differentiate into mature hepatocytes is low among alcoholic hepatitis patients, in whom LR was damaged. Aravinthan et al. discovered [73] that generally diffused inflammation and hepatocyte death occurred in liver fibrosis, and when the telomeres of hepatocyte were shortened, the proportion of aging hepatocytes arrested at the G1/S stage increased. The current opinion is that liver fibrosis is the reason for impaired LR. However, we believe that it is not a simple causal relationship. In livers with chronic injury, fibrosis activates HSCs and HPCs to promote LR reaction, and the transient remodeling of the ECM is necessary for LR. However, various chronic stimulating factors eliminate the termination of ECM remodeling, and persistent activated HSCs produce excessive collagen and elastin fibers to form the network. Finally, the progressive network surrounds and replaces functional liver parenchyma, resulting in the failure of LR and functional degradation of the liver [74]. When blocking the Notch pathways in the HPC niche or applying angiotensin receptor blocker anti-fibrosis treatment to reduce the activation of HSCs and remolding of the ECM, HPCs were more effective at differentiating into hepatocytes to accelerate LR [75]. Cordero-Espinoza and Huch suggested that anti-hepatitis treatment could alleviate fibrosis through balancing tissue regeneration, demonstrating that there is indeed the internal mechanism of sclerosis recession in the liver [74]. We believe that liver fibrosis and LR are interdependent processes. The imbalanced LR leads to liver fibrosis. Moreover, various pathological factors that can lead to liver fibrosis also stimulate the initiation of LR. Time is quite important in this process, and knowing the appropriate time for intervention is key to getting the expected clinical outcomes.
STEATOSIS:
Liver steatosis is defined as an excessive accumulation of triglycerides in hepatocytes. Garnol et al. reported that LR was not affected in a rat model of 70% hepatectomy, in which the animals had diet-induced simple fatty liver degeneration [76]. Matsumoto found that different positions of the autophagosome in the fatty liver of mice were correlated with LR damage [77]. Although whether simple fatty degeneration will damage LR remains controversial, most studies show that severe liver steatosis or fatty liver disease leads to defective LR. At present, the global morbidity of fatty liver disease is increasing, and the damage of LR in patients with steatosis has become a more important clinical problem. Gentric et al. found that there was a defect in EGFR expression in a steatosis rodent model, which suppressed cell cycle progression and delayed the mitosis of hepatocytes [78]. After applying exogenous growth hormone or antioxidant treatment, the normal cell cycle in hepatocytes is restored, revealing that abnormal oxidative stress in the fatty liver may be the important factor hindering LR. Li found that mitochondrial impairment in fatty liver disease led to poor LR and that transplanting apolipoprotein A-1 could accelerate the regeneration of small-for-size fatty liver grafts in mice through enhancing mitochondrial function [79]. Alvarez-Sola et al. developed the fusion molecule fibapo, which includes fibroblast growth factor-19 and apolipoprotein A-1. The application of fibapo before surgery helps regulate metabolism and promote the regeneration of fatty liver [80].
PARTIAL HEPATECTOMY:
In hepatectomy-induced LR, resected volume is closely correlated with the pattern of regeneration. In 90% hepatectomy models, the limited remained resources first meet the liver metabolic function to maintain the homeostasis of the body, resulting in the delay of LR [81]. For 70% hepatectomy models, traditional opinion suggests that almost all residual hepatocytes re-enter the cell cycle for mitosis. However, this is not the case. The adult human liver is comprised of more than 20% polyploid cells, while the number of polyploid cells is as high as 70% in rodents. After PH, polyploid hepatocytes achieve the increase in liver mass through hypertrophy [82]. Many studies have suggested that the volume of LR is significantly negatively correlated with future remnant liver volume (FRLV). However, the removal of a larger volume of liver may cause an increase in the release of mitogens such as TNF-α and IL-6 [83]. Pagano et al. reported that the imbalance between the remaining liver’s function and the body’s metabolic requirement became the most important factor in the regulation of LR. After determining the safe boundary of FRLV to avoid “small liver syndrome” and other related complications, a lower FRLV can promote LR [84].
AGING:
It is generally considered that advanced age has an adverse effect on LR. Zhu et al. reported that the LR rate in 26 elderly patients at 6 months after PH was remarkably lower than that in young patients [85]. Based on their study in 198 patients, Imamura et al. suggested that the aging liver would cause impaired liver function and poor LR after transplantation; therefore, donor age should be considered in the case of liver transplantation [86]. The study of Russolillo et al. included 120 patients who underwent PH, and the results showed that elderly patients had a higher risk of hepatic failure after PH than did younger patients, but the level of LR was insignificantly correlated with age. Consequently, advanced age is not an absolute contraindication for PH; however, precise preoperative assessments are necessary [87]. The decline in LR capacity among elderly patients is caused by multiple interacting extracellular and intracellular factors. The reduction in the autophagy program gives rise to the chronic release of proinflammatory factors by the aging hepatocytes, thereby mediating more severe oxidative stress [88]. Due to the influence of aging on metabolism, HSCs enrich lipid droplets, which have an inadequate ability of promoting LR [89]. HSCs in the aging liver can induce neutrophil infiltration through chemokine ligand 7, and neutrophils perform to produce an excessive oxidative reaction to suppress HPC activation. However, the declined HPC activity may also be a protective mechanism in response to potential tumor risk [90]. Chen et al. used fumarylacetoacetate hydrolase gene-knockout mice to investigate changes in ECM composition in the aging liver. They found that liver structure was loosened and degenerated, which provided the space for the cell transplantation, indicating that aging accumulated during liver disease progression was the precondition for the successful application of cell transplantation therapy [91].
PLATELET COUNT:
It was discovered that after resecting 90% of the liver in mice, the group with a higher platelet count had a higher survival rate, while LR was obviously delayed when thrombocytopenia occurred [92]. Numerous studies have demonstrated that the low platelet count following PH or live liver transplantation is related to postoperative liver dysfunction and death [93–95]. Han et al. discovered that platelet transfusion could result in better LR [96]. It was believed that platelets could rapidly aggregate at the injured site and release the stored mitogens to enhance the regenerative reaction. However, the α-granules released by the platelets contain both pro-proliferation factors and anti-proliferation growth factors, suggesting the multiple effects of platelets on LR. The current explanation is that the platelet-derived growth factors are stored in different α-granules, while the perioperative hepatic hemodynamics determine the selective release of specific α-granules. The secretion of α-granules, rather than the platelet count itself, is correlated with postoperative liver dysfunction and its clinical manifestations [97]. Kirschbaum et al. found that the platelets recruited by PH-induced inflammation can stimulate the proliferation of LSEC and angiogenesis, and further stimulate the LR through activating the AKT and ERK1/2 signaling pathways [98,99]. A recent study proposed that platelets could transfer their RNA to hepatocytes to promote proliferation [100]. However, platelet transfusion also increases the risk of thrombotic events. When trying to promote LR by using platelet-based therapy, we should consider the comprehensive effects and reduce the potential adverse effects.
NEURAL REGULATION AND HORMONES:
Previous studies have discovered that there is a direct feedback relationship between the liver and the brain via the autonomic nervous system [101]. Izumi et al. discovered that LR slowed down when the hepatic branches of the vagus nerve were resected in PH rat models [102]. The mechanism functions when the excessive vagus signal activates macrophages by acetylcholine, and activated macrophages could promote LR through the IL-6/STAT3/FoxM1 pathway. Although only sparse vagus can be observed around the portal vein zone in the liver, the precise multistep regulation constituted by neural cells, immunocytes, and hepatocytes allows the amplification and transmission of an emergency regeneration signal in the liver. Therefore, it can rapidly and precisely regulate LR initiation in the case of acute injury and achieve the survival of the organism. Another study discovered that nerve signals contribute to the release of serotonin in the enterochromaffin cells after PH, thereby facilitating LR indirectly. This suggests that the relay ganglions and organs are vital [103]. Ishtiaq et al. discovered that stress-activated sympathetic nerves induce the elevation of adreno-cortico-tropic hormone and glucocorticoid levels, activate the JAK/ATAT3/NF-κB/IL-6 pathway, and promote LR. At the same time, increased norepinephrine in the plasma can activate the HGF/C-Met and EGF/EGFR pathways in the hepatocytes and antagonize TGF-β in the early stage of LR [104]. Triiodothyronine (T3) hormone can promote mitosis through mediating cyclin D1 in LR [105]. Similar to T3, the thyroid hormone receptor-β-selective agonist can activate Wnt/β-catenin/cyclin D1 signaling to induce hepatocyte proliferation [106].
BA AND GUT MICROBIOTA:
BA is an important signal regulator during the LR process, which can participate in regulating LR through multiple pathways. In patients with obstructive jaundice, LR is suppressed after surgery due to the upregulated expression of TGF-β1, while persistent biliary extra-drainage is also bad for LR because of the excessive loss of BA [107]. Such dual functions of promoting and repressing LR indicate that an appropriate concentration of BA is necessary for the homeostasis of LR. Theoretically, the increased concentration of BA caused by PH will initiate the apoptosis and necrosis in hepatocytes. However, the regulation of the BA receptor prevents the above conditions [108]. BA receptors mainly include nuclear receptor farnesoid X receptor (FXR) and membrane receptor G protein-coupled bile acid receptor 1 (GPBAR1/TGR5). Through a negative feedback mechanism, these 2 BA receptors may synergistically act on the regeneration process and protect the residual liver. FXR can regulate the expression of some important cell cycle transcription factors, such as Foxm1b and cyclin D1, and the FXR-FGF15/19-FGFR4-Hippo signaling pathway could benefit liver growth by preventing BA-induced injury [109,110]. TGR5 exists on the surface of bile duct cells, endothelial cells, and Kupffer cells, but it is not expressed on the hepatocyte membrane surface, which may be related to the prevention of BA overload. BA can activate TGR5, stimulate endothelial cells to produce nitric oxide, and thereby regulate the LR. TGR5 knockout mice manifested cholestasis and LR damage after PH [111]. The portal vein can supply 70% of blood in the liver and carries enterogenic products including endotoxin and bacterial components. The gut microbiota refers to the 100 trillion bacteria existing in the human gastrointestinal tract, and their important effect on regulating liver homeostasis and LR is largely neglected. Wu et al. discovered that in a PH mice model in which animals received antibiotic therapy (ampicillin), changes of the gut microbiota resulted in the loss of tolerance of Kupffer cells. The over-activated Kupffer cells damaged LR through the IL-12/NKT cells/interferon-γ axis [112]. In another case, 90% hepatectomy in rats induced excessively high portal vein and systemic endotoxin levels. When gentamicin was used, the excessive lipopolysaccharide level was reduced, LR was recovered, and the rat survival rate increased from 24% to 56% [113]. In addition to regulating LR directly, gut microbiota also constitutes the intestine-liver axis with BA. The intestine-liver axis describes the bidirectional crosstalk between intestinal nutrient absorption and liver synthetic metabolism. Gut microbiota can regulate BA synthesis, metabolism, and the affinity with BA receptors. On the other hand, BA also provides different patterns of microenvironment for gut microbiota, thus inducing distinct microbial characteristics [114–116]. Consequently, any factor that affects BA composition or gut microbial diversity can markedly affect liver function and regeneration.
Conclusions
We have extensive understanding of the widely discussed and marvelous topic of LR. Nonetheless, we should acknowledge that there are still numerous challenges in gaining a full understand of LR ahead. Here, we illustrated that hepatocytes, BECs, HPCs, and HLSCs could all serve as cell sources for LR. Cell plasticity is a characteristic of LR, and the level of damage and types of different intrahepatic background determine the most suitable cell types for LR. We believe that liver fibrosis and LR are not in a simple causal relationship, but they are interdependent processes. The fibrosis process activates HSCs and HPCs to induce the LR reaction in the liver with chronic injury. Moreover, imbalanced LR can also lead to the occurrence of liver fibrosis. Correcting the abnormal oxidative stress and disturbance of metabolism in the fatty liver may be helpful for promoting regeneration in fatty liver. Other factors such as PH, aging, platelet counts, nerves, hormones, BA, and gut microbiota are crucial targets for regulating LR and need to be investigated further. Recent efforts have focused on shifting from basic research of LR to clinical application. We believe that exploring the mysteries of LR can provide more strategies for solving clinical problems.
Figures
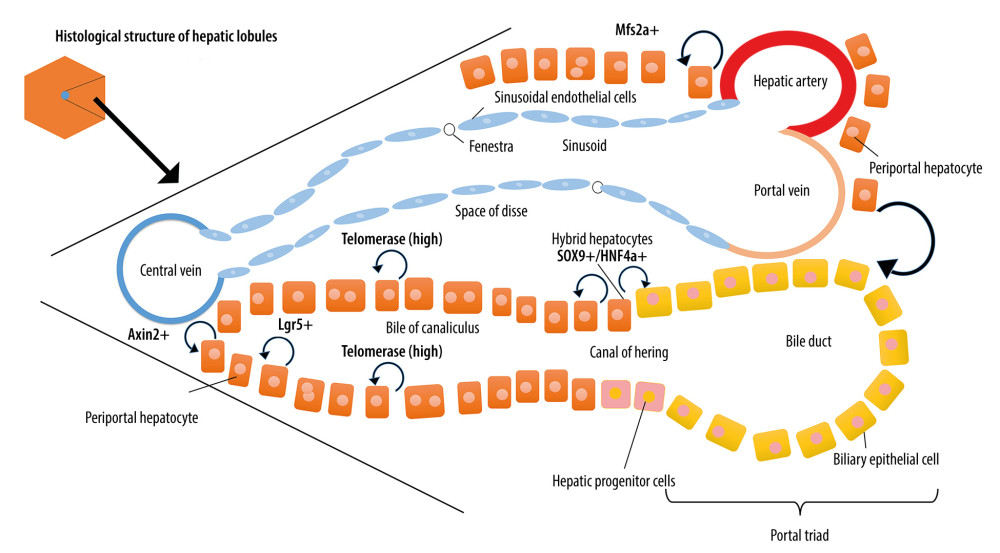 Figure 1. The schematic overview of hepatocyte-mediated liver regeneration (LR). In hepatic lobules, several types of mature hepatocytes have the characteristics of high replication capacity and plasticity. Axin2+ hepatocytes located around the central vein had stem cell-like characteristics; Mfsd2a+ hepatocytes in the portal vein triad region are low in quantity but can be activated, apparently, upon PH or chronic liver injury; a portion of hepatocytes surrounding the portal vein can directly differentiate into biliary cells; Sox9+/HNF4α+ hybrid hepatocytes are located near the branches of the portal vein and terminal biliary tract and can become hepatocytes and BECs after liver injury; Lgr5+ pericentral hepatocytes have a long life and contribute to the homeostasis of the liver; hepatocytes marked by telomerase expression are the main source of hepatocytes in normal liver homeostasis and injury-induced LR.
Figure 1. The schematic overview of hepatocyte-mediated liver regeneration (LR). In hepatic lobules, several types of mature hepatocytes have the characteristics of high replication capacity and plasticity. Axin2+ hepatocytes located around the central vein had stem cell-like characteristics; Mfsd2a+ hepatocytes in the portal vein triad region are low in quantity but can be activated, apparently, upon PH or chronic liver injury; a portion of hepatocytes surrounding the portal vein can directly differentiate into biliary cells; Sox9+/HNF4α+ hybrid hepatocytes are located near the branches of the portal vein and terminal biliary tract and can become hepatocytes and BECs after liver injury; Lgr5+ pericentral hepatocytes have a long life and contribute to the homeostasis of the liver; hepatocytes marked by telomerase expression are the main source of hepatocytes in normal liver homeostasis and injury-induced LR. 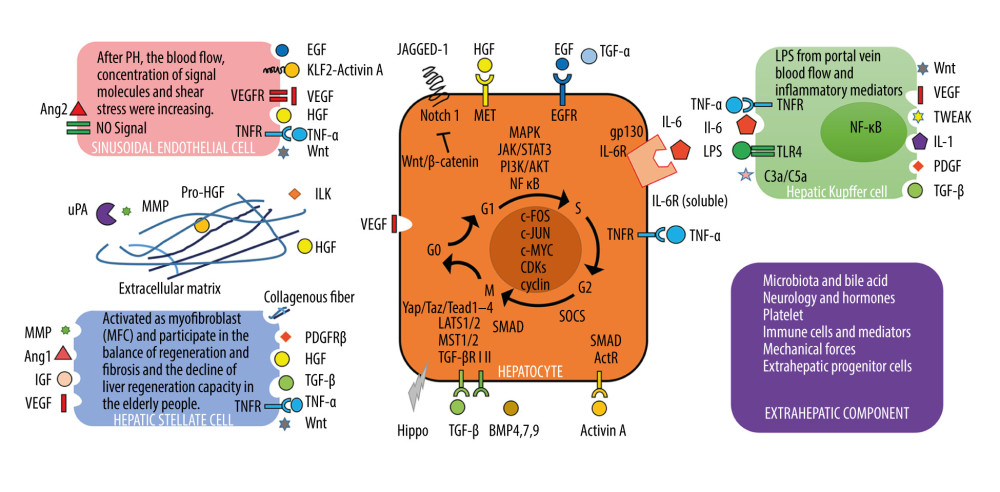 Figure 2. Cells and molecules involved in hepatocyte-mediated liver regeneration. In the hepatocyte-mediated regeneration process, several cells including biliary epithelial cells, Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and extrahepatic cells interact. Additionally, the blood flow stress signal, immune factors, nerves, hormones, bile acid, and microbiota are also involved.
Figure 2. Cells and molecules involved in hepatocyte-mediated liver regeneration. In the hepatocyte-mediated regeneration process, several cells including biliary epithelial cells, Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and extrahepatic cells interact. Additionally, the blood flow stress signal, immune factors, nerves, hormones, bile acid, and microbiota are also involved. 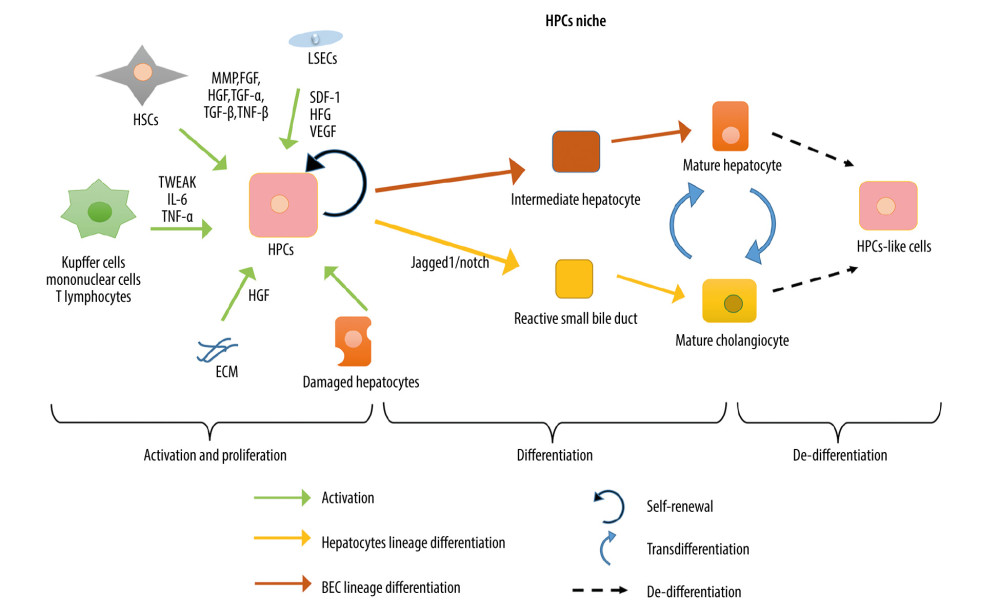 Figure 3. Schematic diagram of “HPCs niche”. The activation, proliferation, differentiation, and de-differentiation of hepatic progenitor cells (HPCs) in “HPCs niche”.
Figure 3. Schematic diagram of “HPCs niche”. The activation, proliferation, differentiation, and de-differentiation of hepatic progenitor cells (HPCs) in “HPCs niche”. 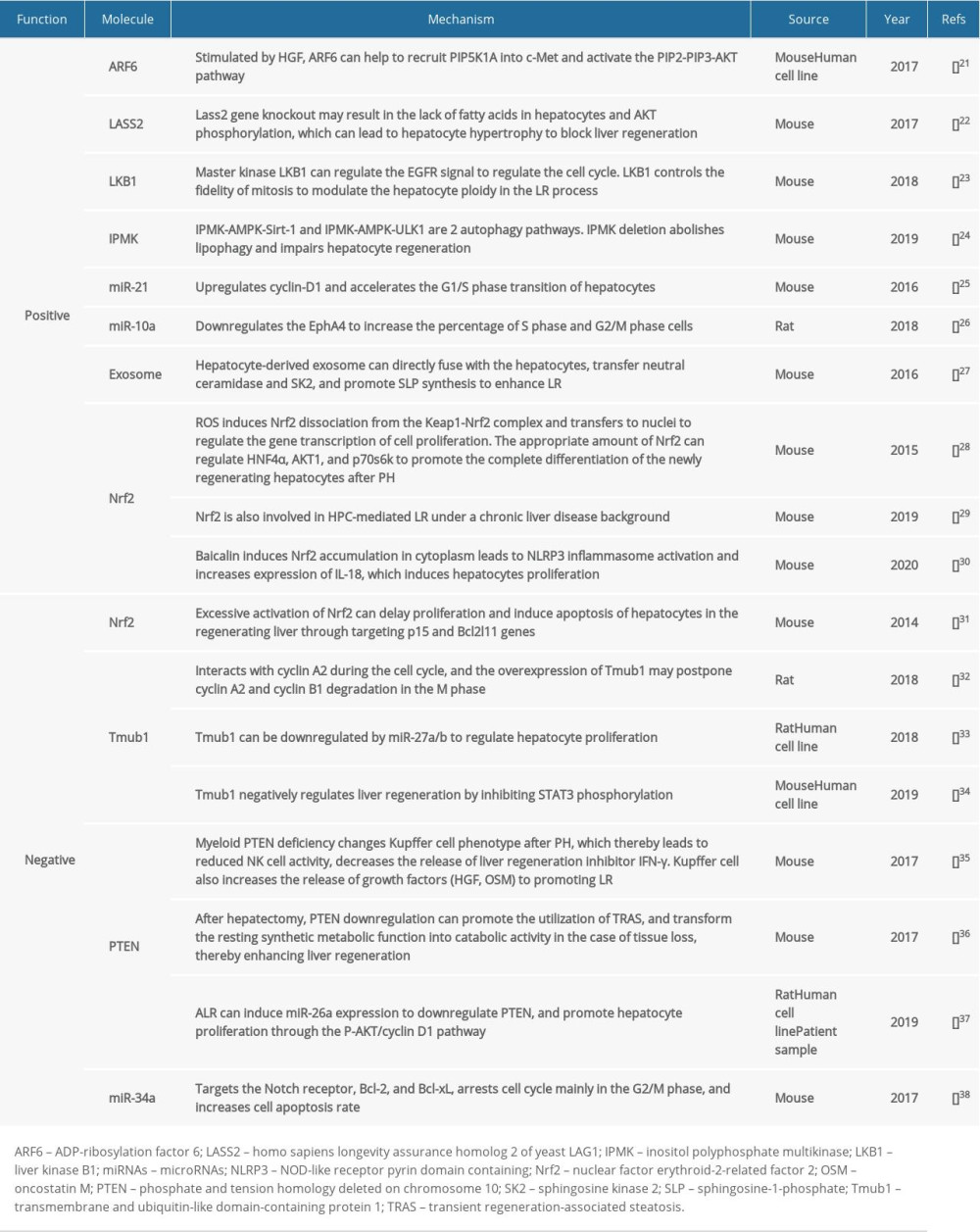
References
1. Preziosi ME, Monga SP, Update on the mechanisms of liver regeneration: Semin Liver Dis, 2017; 37; 141-51
2. Van Haele M, Snoeck J, Roskams T, Human liver regeneration: An etiology dependent process: Int J Mol Sci, 2019; 20; 2332
3. Michalopoulos GK, Hepatostat: Liver regeneration and normal liver tissue maintenance: Hepatology, 2017; 65; 1384-92
4. Wang B, Zhao L, Fish M, Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver: Nature, 2015; 524; 180-85
5. Pu W, Zhang H, Huang X, Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration: Nat Commun, 2016; 7; 13369
6. Schaub JR, Huppert KA, Kurial SNT: Nature, 2018; 557; 247-51
7. Li D, Li W, Hui L, Hybrid hepatocyte: A newly identified player for regeneration in hepatic injuries: Hepatology, 2016; 64; 2244-46
8. Font-Burgada J, Shalapour S, Ramaswamy S, Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer: Cell, 2015; 162; 766-79
9. Ang CH, Hsu SH, Guo F, Lgr5 + pericentral hepatocytes are self-maintained in normal liver regeneration and susceptible to hepatocarcinogenesis: Proc Natl Acad Sci USA, 2019; 116; 19530-40
10. Lin S, Nascimento EM, Gajera CR, Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury: Nature, 2018; 556; 244-48
11. López-Luque J, Fabregat I, Revisiting the liver: From development to regeneration-what we ought to know!: Int J Dev Biol, 2018; 62; 441-451
12. Russell JO, Monga SP, Wnt/β-Catenin signaling in liver development, homeostasis, and pathobiology: Annu Rev Pathol, 2018; 13; 351-78
13. Tao Y, Wang M, Chen E, Tang H, liver regeneration: Analysis of the main relevant signaling molecules: Mediators Inflamm, 2017; 2017 4256352
14. Modares N, Polz R, Haghighi F, IL-6 trans-signaling controls liver regeneration after partial hepatectomy: Hepatology, 2019; 70; 2075-91
15. Hu J, Srivastava K, Wieland M, Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat: Science, 2014; 343; 416-19
16. Rao S, Zaidi S, Banerjee J, Transforming growth factor-β in liver cancer stem cells and regeneration: Hepatol Commun, 2017; 1; 477-93
17. Khan GM, Ghosh A, Variya B, Hepatocyte growth control by SOCS1 and SOCS3: Cytokine, 2019; 121; 154733
18. Lu L, Finegold MJ, Johnson RL, Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration: Exp Mol Med, 2018; 50; e423
19. Zanconato F, Cordenonsi M, Piccolo S, YAP/TAZ at the roots of cancer: Cancer Cell, 2016; 29; 783-803
20. Liu F, Lagares D, Choi KM, Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis: Am J Physiol Lung Cell Mol Physiol, 2015; 308; L344-57
21. Tsai MT, Katagiri N, Ohbayashi N, Regulation of HGF-induced hepatocyte proliferation by the small GTPase Arf6 through the PIP2-producing enzyme PIP5K1A: Sci Rep, 2017; 1; 9438
22. Jin H, Wang C, Gu D, Liver-specific deletion of LASS2 delayed regeneration of mouse liver after partial hepatectomy: Biochem Biophys Res Commun, 2017; 493; 1176-83
23. Maillet V, Boussetta N, Leclerc J, LKB1 as a gatekeeper of hepatocyte proliferation and genomic integrity during liver regeneration: Cell Rep, 2018; 22; 1994-2005
24. Guha P, Tyagi R, Chowdhury S, IPMK mediates activation of ulk signaling and transcriptional regulation of autophagy linked to liver inflammation and regeneration: Cell Rep, 2019; 26; 2692-703
25. Chen X, Song M, Chen W, MicroRNA-21 contributes to liver regeneration by targeting PTEN: Med Sci Monit, 2016; 22; 83-91
26. Luo L, Yu ZP, Qin H, Exosomal MicroRNA-10a is associated with liver regeneration in rats through downregulation of EphA4: Chin Med J (Engl), 2018; 131; 454-60
27. Nojima H, Freeman CM, Schuster RM, Hepatocyte exosomes mediate liver repair and regeneration via sphingosine-1-phosphate: J Hepatol, 2016; 64; 60-68
28. Morales-González JA, Madrigal-Santillán E, Morales-González Á, what is known regarding the participation of factor Nrf-2 in liver regeneration?: Cells, 2015; 4; 169-77
29. Fragoulis A, Schenkel J, Herzog M, Nrf2 ameliorates DDC-induced sclerosing cholangitis and biliary fibrosis and improves the regenerative capacity of the liver: Toxicol Sci, 2019; 169; 485-98
30. Shi L, Zhang S, Huang Z, Baicalin promotes liver regeneration after acetaminophen-induced liver injury by inducing NLRP3 inflammasome activation: Free Radic Biol Med, 2020 [Online ahead of print]
31. Köhler UA, Kurinna S, Schwitter D, Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis: Hepatology, 2014; 60; 670-78
32. Fu H, Xu J, Chen J, Li G, Microarray analysis reveals Tmub1 as a cell cycle-associated protein in rat hepatocytes: Mol Med Rep, 2018; 17; 4337-44
33. Lan X, Li G, Liu H, MiR-27a/b regulates liver regeneration by posttranscriptional modification of Tmub1: Dig Dis Sci, 2018; 63; 2362-72
34. Fu H, Dong R, Zhang Y, Tmub1 negatively regulates liver regeneration via inhibiting STAT3 phosphorylation: Cell Signal, 2019; 55; 65-72
35. Ma WT, Jia YJ, Liu QZ, Modulation of liver regeneration via myeloid PTEN deficiency: Cell Death Dis, 2017; 8; e2827
36. Kachaylo E, Tschuor C, Calo N, PTEN down-regulation promotes β-oxidation to fuel hypertrophic liver growth after hepatectomy in mice: Hepatology, 2017; 66; 908-21
37. Gupta P, Sata TN, Ahamad N, Augmenter of liver regeneration enhances cell proliferation through the microRNA-26a/Akt/cyclin D1 pathway in hepatic cells: Hepatol Res, 2019; 49; 1341-52
38. Wang XP, Zhou J, Han M, MicroRNA-34a regulates liver regeneration and the development of liver cancer in rats by targeting Notch signaling pathway: Oncotarget, 2017; 8; 13264-76
39. Paranjpe S, Bowen WC, Mars WM, Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation: Hepatology, 2016; 64; 1711-24
40. Bloise E, Ciarmela P, Dela Cruz C, Activin A in mammalian physiology: Physiol Rev, 2019; 99; 739-80
41. de Jong IEM, Matton APM, van Praagh JB, Peribiliary glands are key in regeneration of the human biliary epithelium after severe bile duct injury: Hepatology, 2019; 69; 1719-34
42. Deng X, Zhang X, Li W, chronic liver injury induces conversion of biliary epithelial cells into hepatocytes: Cell Stem Cell, 2018; 23; 114-22
43. Raven A, Lu WY, Man TY, Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration: Nature, 2017; 547; 350-54
44. Choi TY, Ninov N, Stainier DY, Shin D, Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish: Gastroenterology, 2014; 146; 776-88
45. Popper H, Kent G, Stein R, Ductular cell reaction in the liver in hepatic injury: J Mt Sinai Hosp NY, 1957; 24; 551-56
46. Van Haele M, Roskams T, Hepatic progenitor cells: an update: Gastroenterol Clin North Am, 2017; 46; 409-20
47. Tirnitz-Parker JEE, Forbes SJ, Olynyk JK, Ramm GA, cellular plasticity in liver regeneration: Spotlight on cholangiocytes: Hepatology, 2019; 69; 2286-89
48. Bria A, Marda J, Zhou J, Hepatic progenitor cell activation in liver repair: Liver Res, 2017; 1; 81-87
49. Dezső K, Nagy P, Paku S, Human liver regeneration following massive hepatic necrosis: Two distinct patterns: J Gastroenterol Hepatol, 2020; 35; 124-34
50. Tarlow BD, Pelz C, Naugler WE, Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes: Cell Stem Cell, 2014; 15; 605-18
51. He J, Lu H, Zou Q, Luo L, Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish: Gastroenterology, 2014; 146; 789-800
52. Lu WY, Bird TG, Boulter L, Hepatic progenitor cells of biliary origin with liver repopulation capacity: Nat Cell Biol, 2015; 17; 971-83
53. Zheng YJ, Pan MX, Wang YClinical significance and correlation of ductular reaction in hepatobiliary diseases: Zhonghua Gan Zang Bing Za Zhi, 2018; 26; 637-40 [in Chinese]
54. Liu WH, Ren LN, Wang T, The involving roles of intrahepatic and extrahepatic stem/progenitor cells (SPCs) to liver regeneration: Int J Biol Sci, 2016; 12; 954-63
55. Shang H, Wang Z, Song Y, Liver progenitor cells-mediated liver regeneration in liver cirrhosis: Hepatol Int, 2016; 10; 440-47
56. Kitade M, Kaji K, Yoshiji H, Relationship between hepatic progenitor cell-mediated liver regeneration and non-parenchymal cells: Hepatol Res, 2016; 46; 1187-93
57. Boulter L, Govaere O, Bird TG, Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease: Nat Med, 2012; 18; 572-79
58. Campagnoli C, Roberts IA, Kumar S, Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow: Blood, 2001; 98; 2396-402
59. Herrera MB, Bruno S, Buttiglieri S, Isolation and characterization of a stem cell population from adult human liver: Stem Cells, 2006; 24; 2840-50
60. Najimi M, Khuu DN, Lysy PA, Adult-derived human liver mesenchymal-like cells as a potential progenitor reservoir of hepatocytes?: Cell Transplant, 2007; 16; 717-28
61. Pan Q, Fouraschen SMG, Kaya FSFA, Mobilization of hepatic mesenchymal stem cells from human liver grafts: Liver Transpl, 2011; 17; 596-609
62. Lee JH, Park HJ, Kim YA, The phenotypic characteristic of liver-derived stem cells from adult human deceased donor liver: Transplant Proc, 2012; 44; 1110-12
63. Luo X, Gupta K, Ananthanarayanan A, Directed differentiation of adult liver derived mesenchymal like stem cells into functional hepatocytes: Sci Rep, 2018; 8; 2818
64. Li J, Xin J, Zhang L, Human hepatic progenitor cells express hematopoietic cell markers CD45 and CD109: Int J Med Sci, 2013; 11; 65-79
65. Wang Y, Yu X, Chen E, Li L, Liver-derived human mesenchymal stem cells: A novel therapeutic source for liver diseases: Stem Cell Res Ther, 2016; 7; 71
66. Kholodenko IV, Kurbatov LK, Kholodenko RV, Mesenchymal stem cells in the adult human liver: Hype or hope?: Cells, 2019; 8; 1127
67. Duret C, Gerbal-Chaloin S, Ramos J, Isolation, characterization, and differentiation to hepatocyte-like cells of nonparenchymal epithelial cells from adult human liver: Stem Cells, 2007; 25; 1779-90
68. Lee JH, Park HJ, Kim YA, Differentiation and major histocompatibility complex antigen expression in human liver-derived stem cells: Transplant Proc, 2012; 44; 1113-15
69. Herrera MB, Fonsato V, Bruno S, Human liver stem cells improve liver injury in a model of fulminant liver failure: Hepatology, 2013; 57; 311-19
70. Lee J, Choi J, Kang S, Hepatogenic potential and liver regeneration effect of human liver-derived mesenchymal-like stem cells: Cells, 2020; 9; 1521
71. Fouraschen SMG, Pan Q, de Ruiter PE, Secreted factors of human liver-derived mesenchymal stem cells promote liver regeneration early after partial hepatectomy: Stem Cells Dev, 2012; 21; 2410-19
72. Dubuquoy L, Louvet A, Lassailly G, Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis: Gut, 2015; 64; 1949-60
73. Aravinthan A, Scarpini C, Tachtatzis P, Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease: J Hepatol, 2013; 58; 549-56
74. Cordero-Espinoza L, Huch M, The balancing act of the liver: Tissue regeneration versus fibrosis: J Clin Invest, 2018; 128; 85-96
75. Kitade M, Kaji K, Nishimura N, Blocking development of liver fibrosis augments hepatic progenitor cell-derived liver regeneration in a mouse chronic liver injury model: Hepatol Res, 2019; 49; 1034-45
76. Garnol T, Kučera O, Staňková P, Does simple steatosis affect liver regeneration after partial hepatectomy in rats?: Acta Medica (Hradec Kralove), 2016; 59; 35-42
77. Matsumoto Y, Yoshizumi T, Toshima T, Ectopic localization of autophagosome in fatty liver is a key factor for liver regeneration: Organogenesis, 2019; 15; 24-34
78. Gentric G, Maillet V, Paradis V, Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease: J Clin Invest, 2015; 125; 981-92
79. Li CX, Chen LL, Li XC, ApoA-1 accelerates regeneration of small-for-size fatty liver graft after transplantation: Life Sci, 2018; 215; 128-35
80. Alvarez-Sola G, Uriarte I, Latasa MU, Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration: Gut, 2017; 66; 1818-28
81. Kholodenko IV, Yarygin KN, Cellular mechanisms of liver regeneration and cell-based therapies of liver diseases: Biomed Res Int, 2017; 2017 8910821
82. Wang MJ, Chen F, Lau JTY, Hu YP, Hepatocyte polyploidization and its association with pathophysiological processes: Cell Death Dis, 2017; 8; e2805
83. Pagano D, Spada M, Parikh V, Liver regeneration after liver resection: Clinical aspects and correlation with infective complications: World J Gastroenterol, 2014; 20; 6953-60
84. Pagano D, Gruttadauria S, Impact of future remnant liver volume on post-hepatectomy regeneration in non-cirrhotic livers: Front Surg, 2014; 1; 10
85. Zhu C, Ikemoto T, Utsunomiya T, Senescence-related genes possibly responsible for poor liver regeneration after hepatectomy in elderly patients: J Gastroenterol Hepatol, 2014; 29; 1102-8
86. Imamura H, Hidaka M, Soyama A, A donor age-based and graft volume-based analysis for living donor liver transplantation in elderly recipients: Transplant Direct, 2017; 3; e168
87. Russolillo N, Ratti F, Viganò L, The influence of aging on hepatic regeneration and early outcome after portal vein occlusion: A case-control study: Ann Surg Oncol, 2015; 22; 4046-51
88. Pibiri M, Liver regeneration in aged mice: New insights: Aging (Albany NY), 2018; 10; 1801-24
89. Saito Y, Morine Y, Shimada M, Mechanism of impairment on liver regeneration in elderly patients: Role of hepatic stellate cell function: Hepatol Res, 2017; 47; 505-13
90. Cheng Y, Wang X, Wang B, Aging-associated oxidative stress inhibits liver progenitor cell activation in mice: Aging (Albany NY), 2017; 9; 1359-74
91. Chen J, Yang T, Song S, Senescence suppressed proliferation of host hepatocytes is precondition for liver repopulation: Biochem Biophys Res Commun, 2019; 516; 591-98
92. Kurokawa T, Ohkohchi N, Platelets in liver disease, cancer and regeneration: World J Gastroenterol, 2017; 23; 3228-39
93. Mehrabi A, Golriz M, Khajeh E, Meta-analysis of the prognostic role of perioperative platelet count in posthepatectomy liver failure and mortality: Br J Surg, 2018; 105; 1254-61
94. Beltrame P, Rodriguez S, Brandão ABM, Low platelet count: Predictor of death and graft loss after liver transplantation: World J Hepatol, 2019; 11; 99-108
95. Takahashi K, Kurokawa T, Oshiro Y, Postoperative decrease in platelet counts is associated with delayed liver function recovery and complications after partial hepatectomy: Tohoku J Exp Med, 2016; 239; 47-55
96. Han S, Park HW, Song JH, Association between intraoperative platelet transfusion and early graft regeneration in living donor liver transplantation: Ann Surg, 2016; 264; 1065-72
97. Starlinge P, Haegele S, Offensperger F, The profile of platelet α-granule released molecules affects postoperative liver regeneration: Hepatology, 2016; 63; 1675-88
98. Kirschbaum M, Jenne CN, Veldhuis ZJ, Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice: Liver Int, 2017; 37; 1731-37
99. Aryal B, Yamakuchi M, Shimizu T, Therapeutic implication of platelets in liver regeneration-hopes and hues: Expert Rev Gastroenterol Hepatol, 2018; 12; 1219-28
100. Kirschbaum M, Karimian G, Adelmeijer J, Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation: Blood, 2015; 126; 798-806
101. Kamimura K, Inoue R, Nagoya T, Autonomic nervous system network and liver regeneration: World J Gastroenterol, 2018; 24; 1616-21
102. Izumi T, Imai J, Yamamoto J, Vagus-macrophage-hepatocyte link promotes post-injury liver regeneration and whole-body survival through hepatic FoxM1 activation: Nat Commun, 2018; 9; 5300
103. Inoue R, Kamimura K, Nagoya T, Effect of a neural relay on liver regeneration in mice: Activation of serotonin release from the gastrointestinal tract: FEBS Open Bio, 2018; 8; 449-60
104. Ishtiaq SM, Khan JA, Arshad MI, Psychosocial-stress, liver regeneration and weight gain: A conspicuous pathophysiological triad: Cell Physiol Biochem, 2018; 46; 1-8
105. Fanti M, Singh S, Ledda-Columbano GM, Tri-iodothyronine induces hepatocyte proliferation by protein kinase A-dependent β-catenin activation in rodents: Hepatology, 2014; 59; 2309-20
106. Alvarado TF, Puliga E, Preziosi M, Thyroid hormone receptor β agonist induces β-catenin-dependent hepatocyte proliferation in mice: Implications in hepatic regeneration: Gene Expr, 2016; 17; 19-34
107. Otao R, Beppu T, Isiko T, External biliary drainage and liver regeneration after major hepatectomy: Br J Surg, 2012; 99; 1569-74
108. Merlen G, Ursic-Bedoya J, Jourdainne V, Bile acids and their receptors during liver regeneration: “Dangerous protectors”: Mol Aspects Med, 2017; 56; 25-33
109. Ji S, Liu Q, Zhang S, FGF15 activates Hippo signaling to suppress bile acid metabolism and liver tumorigenesis: Dev Cell, 2019; 48; 460-74
110. Li G, Guo GL, Farnesoid X receptor, the bile acid sensing nuclear receptor, in liver regeneration: Acta Pharm Sin B, 2015; 5; 93-98
111. Jourdainne V, Péan N, Doignon I, The bile acid receptor TGR5 and liver regeneration: Dig Dis, 2015; 33; 319-26
112. Wu X, Sun R, Chen Y, Oral ampicillin inhibits liver regeneration by breaking hepatic innate immune tolerance normally maintained by gut commensal bacteria: Hepatology, 2015; 62; 253-64
113. Ren W, Wang X, Zhang A, Selective bowel decontamination improves the survival of 90% hepatectomy in rats: J Surg Res, 2015; 195; 454-64
114. Adolph TE, Grander C, Moschen AR, Tilg H, Liver-microbiome axis in health and disease: Trends Immunol, 2018; 39; 712-23
115. Liu HX, Keane R, Sheng L, Wan YJ, Implications of microbiota and bile acid in liver injury and regeneration: J Hepatol, 2015; 63; 1502-10
116. Liu HX, Rocha CS, Dandekar S, Wan YJ, Functional analysis of the relationship between intestinal microbiota and the expression of hepatic genes and pathways during the course of liver regeneration: J Hepatol, 2016; 64(3); 641-50
Figures
Figure 1. The schematic overview of hepatocyte-mediated liver regeneration (LR). In hepatic lobules, several types of mature hepatocytes have the characteristics of high replication capacity and plasticity. Axin2+ hepatocytes located around the central vein had stem cell-like characteristics; Mfsd2a+ hepatocytes in the portal vein triad region are low in quantity but can be activated, apparently, upon PH or chronic liver injury; a portion of hepatocytes surrounding the portal vein can directly differentiate into biliary cells; Sox9+/HNF4α+ hybrid hepatocytes are located near the branches of the portal vein and terminal biliary tract and can become hepatocytes and BECs after liver injury; Lgr5+ pericentral hepatocytes have a long life and contribute to the homeostasis of the liver; hepatocytes marked by telomerase expression are the main source of hepatocytes in normal liver homeostasis and injury-induced LR.Figure 2. Cells and molecules involved in hepatocyte-mediated liver regeneration. In the hepatocyte-mediated regeneration process, several cells including biliary epithelial cells, Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and extrahepatic cells interact. Additionally, the blood flow stress signal, immune factors, nerves, hormones, bile acid, and microbiota are also involved.Figure 3. Schematic diagram of “HPCs niche”. The activation, proliferation, differentiation, and de-differentiation of hepatic progenitor cells (HPCs) in “HPCs niche”. In Press
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952