19 January 2022: Review Articles
Role of NLRP3 Inflammasome in Myocardial Ischemia-Reperfusion Injury and Ventricular Remodeling
Shichun Shen1ACE, Zhen Wang1DF, Haozhong Sun1BC, Likun Ma1AFG*DOI: 10.12659/MSM.934255
Med Sci Monit 2022; 28:e934255






Abstract
ABSTRACT: Reperfusion therapy is the optimal therapy for acute myocardial infarction (AMI), but acute inflammatory injury and chronic heart failure (HF) after myocardial ischemia and reperfusion (MI/R) remain the leading cause of death after AMI. Pyroptosis, a newly discovered form of cell death, has been proven to play a significant role in the acute reperfusion process and the subsequent chronic process of ventricular remodeling. Current research shows that multiple stimuli activate the pyroptotic signaling pathway and contribute to cell death and nonbacterial inflammation after MI/R. These stimuli promote the assembly of the nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome by activating NLRP3. The mature NLRP3 inflammasome cleaves procaspase-1 to active caspase-1, which leads to mature processing of interleukin (IL)-18, IL-1β, and gasdermin D (GSDMD) protein. That eventually results in cell lysis and generation of nonbacterial inflammation. The present review summarizes the mechanism of NLRP3 inflammasome activation after MI/R and discusses the role that NLRP3-mediated pyroptosis plays in the pathophysiology of MI/R injury and ventricular remodeling. We also discuss potential mechanisms and targeted therapy for which there is evidence supporting treatment of ischemic heart disease.
Keywords: Myocardial Reperfusion Injury, NLRP3 Protein, Human, Pyroptosis, Ventricular Remodeling, Humans, Inflammasomes, Inflammation, NLR Family, Pyrin Domain-Containing 3 Protein, Signal Transduction
Background
Ischemic heart disease (IHD) is the leading cause of morbidity and mortality worldwide [1]. The leading cause of death from IHD is acute myocardial infarction (AMI). Rupture of the fibrous cap on the surface of an atherosclerotic plaque on the coronary artery wall is the main cause of AMI, and leads to secondary thrombosis, occlusion of the coronary lumen, blood flow obstruction, and stagnation [2]. All these pathological changes lead to a sharp drop in oxygen and energy available for myocardial tissue supplied by the culprit coronary artery; the result is irreversible necrosis of the myocardium [3]. In essence, AMI is an imbalance in oxygen supply and demand [4]. Thus, the most effective strategy for rescuing the dying cardiomyocytes is immediate reperfusion therapy to restore blood flow [5]. However, a series of studies have demonstrated that although reperfusion treatment minimizes the area of infarction caused by hypoxia in infarcted myocardium, it also provokes a series of events that lead to damage to myocardial function, metabolism, and electrophysiology, which is known as myocardial ischemia/reperfusion (MI/R) injury [6].
Whether patients who experience AMI are treated with reperfusion or not, their damaged myocardium is often unable to contract to complete the heart’s pumping function because of irreversible loss of function and activity [7]. To maintain constant cardiac output, fibroblasts become hyperplastic and healthy cardiomyocytes experience hypertrophy and elongation [8]. The site and extent of AMI are the most crucial predictors of ventricular remodeling [9]. Ventricular remodeling is a chronic pathological alteration that contributes to congestive heart failure (HF), which is the long-term cause of death from AMI [10].
Recently, an increasing number of studies have documented the role of pyroptosis in ischemic cardiomyopathy [11–14]. Pyroptosis is a type of programmed cell death that was first observed in macrophages infected by salmonella [15]. The process is mainly dependent on the enzymatic activity of the cysteine-dependent aspartate-specific protease family, which can be activated by the nucleotide-binding and oligomerization (NOD)-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome [16]. The NLRP3 inflammasome is one of the most widely studied inflammasomes. NLRP3 can recruit precursors of caspase-1 and apoptosis-associated speck-like protein that contain a caspase-recruiting domain (CARD) (apoptosis speck-like protein; ASC) to form mature NLRP3 inflammasomes [16]. A series of damaging factors, such as complete pathogens, pathogen-related molecular patterns (PAMPs) with diverse structures, host-derived risk signals (danger-related molecular patterns, DAMPs), and environmental stimuli, can activate NLRP3 to recruit procaspase-1 and ASC to construct a mature NLRP3 inflammasome [17]. The NLRP3 inflammasome is involved in the pathogenesis of many diseases [18–20]. Inhibition of NLRP3-mediated pyroptosis may be an important target of therapy for MI/R injury and ventricular remodeling, and it has a significant impact on the development of and prognosis for AMI [21]. In the present review, we systematically summarize the current molecular mechanism of NLRP3-mediated pyroptosis and highlight its role and therapeutic significance in MI/R and later myocardial remodeling. Our aim is to contribute to advancing the concept of the NLRP3 inflammasome-mediated signaling pathway as a future target for treatment of coronary heart disease.
Activation of the NLRP3 Inflammasome
POTASSIUM EFFLUX:
During acute myocardial ischemia, cellular potassium efflux is increased and cellular potassium influx is decreased in cardiomyocytes. An open ATP-sensitive potassium (KATP) channel is the main mechanism of unidirectional potassium efflux in cardiomyocytes that experience acute ischemia and it is beneficial to accumulation of extracellular potassium [32]. Sodium-activated potassium channels and free fatty acid potassium channels have been identified as related to ischemia-induced potassium efflux [33,34]. ATP hydrolyzes the decrease in phosphorylation level during AMI, simultaneously promoting dysfunction in the sodium-potassium pump, lowering cellular potassium influx [35].
Potassium promotion of activation of IL-1β was first observed in intracellular LPS-activated macrophages [36]. Since then, it has been confirmed that stimulation of the NLRP3 inflammasome by acute ischemia, ATP, nigericin, and crystalline matter can cause intracellular potassium efflux and subsequently activate the NLRP3 inflammasome [32,37]. It is widely recognized that alteration in potassium concentration gradients in cardiomyocytes contributes to activation of the NLRP3 inflammasome [38].
What remains unclear, however, is the role of potassium efflux in activation of the NLRP3 inflammasome and the relationship between potassium efflux and the NLRP3 inflammasome. This is notwithstanding the variation in potassium concentration gradients caused by potassium efflux, which is a consideration as the upstream signal for the NLRP3 inflammasome. Successful assembly of the NLRP3 inflammasome has been observed without the outflow of potassium [39]. Stimuli such as GB111-NH2 and CL097 promote activation of the NLRP3 inflammasome, which is not accompanied by potassium efflux [40]. This finding suggests that another signal cascade downstream from the potassium efflux and other parallel-signaling pathways independent of potassium efflux can regulate activation of the NLRP3 inflammasome. A recent study revealed that release of IL-1β decreases when the intracellular potassium outflow is stopped [41]. When the potassium outflow increases, it directly increases activation of caspase-1, expression of NIMA-related kinase 7 (NEK7), and formation of the NEK7-NLRP3 complex [42]. NEK-7, a serine/threonine-protein kinase that can regulate cell cycle and mitosis, has proven to be a downstream signal for potassium efflux [42]. NEK-1 promotes the activation of NLRP3 inflammasomes by interacting with the LRR domain of NLRP3 to form a NEK7-NLRP3 complex [43]. There is no rationale for potassium efflux activation of caspase-1 and IL-1β after knockout by NEK-7. Studies have revealed that NEK7 is essential for the NLRP3 inflammasome assembly caused by potassium efflux (Figure 2) [44,45].
CALCIUM OVERLOAD:
An increase in intracellular anerobic glycolysis reduces the potential of hydrogen (pH) in the cytoplasm and increases the hydron concentration during AMI. The intracellular hydron is transferred to the outside of the cell through the sodium-hydron exchanger, increasing the intracellular sodium concentration. To balance the intracellular sodium concentration, the sodium-calcium exchanger transports excessive intracellular sodium out of the cell and transports extracellular calcium into the cell, causing a calcium overload [46]. In addition to the transfer of extracellular calcium, calcium released from the intracellular sarcoplasmic reticulum (SR) is another source of calcium overload. SR is the main intracellular calcium reservoir. Acute hypoxia leads to production of a large amount of oxygen free radicals in cardiomyocytes, which damage the SR membrane, triggering release of calcium into the cytoplasm and aggravating calcium overload [47]. After reperfusion, the reduced concentration of extracellular hydron promotes sodium-hydron and sodium-calcium exchange, worsening calcium overload [46].
Calcium is a common secondary messenger in cells that participates in a series of basic cellular events, including autophagy, apoptosis, and inflammation. Some NLRP3 stimuli reportedly can induce variations in calcium concentration [48]. The intracellular calcium concentration increases during NLRP3 inflammasome activation [49]. After inhibiting intracellular calcium increases, activation of the NLRP3 inflammasome is downregulated. The endoplasmic reticulum (ER) is an important calcium storage organelle in the cardiomyocytes and plays a crucial role in calcium signal transduction by releasing calcium into the cytoplasm through the 1, 4, 5-trisphosphate receptor (IP3R) [49]. Phospholipase C downstream of G protein-coupled receptors is activated to mediate the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2), generating 1, 4, 5-triphosphate muscle alcohol (IP3) [50]. IP3 interacts with IP3R to induce calcium outflow from the ER to the cytoplasm. When IP3R is inhibited, calcium mobilization and activation of the NLRP3 inflammasome can be inhibited [51].
How the mechanism of calcium overload in cytoplasm promotes activation of the NLRP3 inflammasome remains unclear. Researchers have observed that with the rise in calcium concentration in the cytoplasm, the level of mature NLRP3 increases [48]. Therefore, the hypothesis is that the NLRP3 inflammasome is activated directly by calcium overload. Meanwhile, calcium flows into the mitochondria through mitochondrial calcium channels and voltage-dependent anion-selective channels, causing mitochondrial instability [52–54]. Cardiolipin and mitochondrial DNA (mtDNA) are released from the mitochondria to activate NLRP3 and promote maturation of caspase-1 and inflammatory factors [55–57]. Another study found that activation of the NLRP3 inflammasome and secretion of IL-1β were significantly inhibited after a calcium chelator was applied [58]. ATP and Leu-Leu-Ome, 2 stimuli of NLRP3, regulate the NLRP3 inflammasome without causing fluctuations in the concentration of calcium in the cytoplasm [59]. That finding indicates that although calcium overload plays an important role in activating the NLRP3 inflammasome, the inflammasome can be activated in a calcium overload-independent manner (Figure 2).
REACTIVE OXYGEN SPECIES:
ROS are the most common second messengers of oxidative stress inside and outside the cell. They participate in a series of regulatory mechanisms inherent in growth and development of cells in the body. An excess of ROS, however, promotes oxidative stress and damages the mitochondria, DNA, and proteins, leading to cell dysfunction [60]. AMI leads to inhibition of activity in the mitochondrial electron transport chain complex and leakage of electrons. Complexes I and III have been regarded as the primary source of ROS [61,62]. An accumulation of redundant succinic acid in cardiomyocytes during hypoxia promotes reverse electron transport, producing ROS throughout the complex I [63]. After myocardial reperfusion, the opening of the mitochondrial permeability transition pore (MPTP) increases the level of ROS, which in turn increases the number of openings in the MPTP, creating a vicious cycle [64]. In addition, nicotinamide adenine dinucleotide phosphate oxidase has been shown to play an important role in ROS production in MI/R. Braunerreuther et al found that after MI/R, mice with specific knockouts for NOX1 and NOX2 had significantly fewer areas of MI than wild-type mice [65]. Matsushima et al. found that in mice, MI/R treatment after knockout of myocardial-specific NOX4 reduced ROS production and MI area [66].
Although ROS are not essential to the cascade for activation of the NLRP3 inflammasomes, they are a crucial upstream signal for activation of the inflammasomes [60,67]. It is worth noting that the process of NLRP3 activation by various NLRP3 stimuli is accompanied by production of ROS [68,69]. ROS can regulate apoptosis through NF-κB signaling [70]. The level of activation of the NLRP3 inflammasome increases in proportion to increases in the NF-κB level. To confirm that ROS triggers activation of the NLRP3 inflammasomes by regulating NF-κB, researchers tested various antioxidants and eventually demonstrated that NF-κB and NLRP3 activation were positively correlated during oxidation or anti-oxidation [71].
However, ROS does not always stimulate an increase in NF-κB. It has been reported that slight increases in ROS are conducive to activation of NF-κB, but excessive accumulation of ROS prevents activation of NF-κB. These results may indicate that NF-κB is not the only signal downstream of ROS that plays a role in activation and assembly of the NLRP3 inflammasomes [72].
Thioredoxin-interacting protein (TXNIP) is the ligand of NLRP3 in the cytoplasm [73]. Under normal physiological conditions, thioredoxin (TRX) inhibits the activity of TXNIP by binding to TXNIP [74]. Transient increases in intracellular ROS levels promote a dissociation between TRX and TXNIP and activation of the NLRP3 inflammasome through increased binding of TXNIP and NLRP3 [75,76]. In the non-canonical caspase-11-mediated signaling pathway, ROS activates NLRP3 by promoting the expression and activation of caspase-11. However, the mechanism of ROS that regulates caspase-11 needs to be further explored (Figure 2) [77].
MITOCHONDRIAL DYSFUNCTION:
During ischemia, the opening of MPTP causes water and cytoplasm to penetrate the mitochondria, resulting in swelling and rupture of the outer mitochondrial membrane and leading to loss of factors and induction of apoptosis in cytochromes. The degree to which MPTP is open can be further aggravated during reperfusion. Some studies have found that inhibiting the opening of MPTP can reduce damage to and death of myocardial cells [78]. The mitochondria are the most important source of ROS in cells and the most crucial organelle for oxidative stress. The mitochondria produce ROS through the oxidative respiratory chain [79]. Electron transport of the mitochondria is continuously damaged during MI. The damage progresses from the distal end of complex I, leading first to a reduction in oxidation of glutamate and succinic acid, which are the substrate of complex I and the donor of complex II, respectively. As the ischemia worsens, complex III and the phosphorylation device, including complex IV and the adenine nucleotide transporters, are damaged, too [80–82]. Rotenone, a mitochondrial complex I inhibitor, can cause loss of mitochondrial membrane potential and a high level of ROS after it is added to the cell [83,84]. A similar but slight effect can be observed in the cell when antimycin A acts on mitochondrial complex II and 2-thenoyltrifluoroacetone acts on complex III [85,86]. These findings indicate that complex I may be the principal site of production of ROS by the mitochondria.
Abnormal calcium mobilization induces mitochondrial dysfunction and can activate the NLRP3 inflammasome [87]. Transport of calcium into the mitochondria can lead to loss of the membrane’s potential to activate the NLRP3 inflammasome [88,89]. NLRP3 stimuli such as ATP, urea microcrystals, and nigericin reportedly can cause the loss of potential in the mitochondrial membrane. ROS-induced release of oxidized mtDNA from the mitochondria can interact with and activate the NLRP3 inflammasome [90,91]. Diphosphatidylglycerol exists on the inner membrane of mitochondria. Damage to the cell can result in transfer of diphosphatidylglycerol from the inner membrane to the outer membrane and binding to the free NLRP3 LRRs in the cytoplasm, promoting activation of NLRP3 (Figure 2) [92].
LYSOSOMAL DAMAGE:
Lysosomes are the organelles that hydrolyze proteins, nucleic acids, polysaccharides, and other macromolecules. Lysosomal damage is widely believed to be related to myocardial ischemia. In the last century, Acosta et al have found that myocardial ischemia increases the instability of lysosomal membranes, releasing powerful hydrolases into the cytoplasm and causing damage to intracellular organelles and important submicroscopic structures [93]. Lysosomes contain multiple types of hydrolase, including cathepsin B. Not only does the lysosome cause decomposition of damaging factors that can permeate the cell, it also swallows internal impaired organelles and useless substances. Particulate foreign substances, such as alum, silica, asbestos, amyloid-beta protein, cholesterol crystals, and calcium crystals, can hurt the lysosome membrane and promote the release of cathepsin B [94]. The amount of cathepsin B released into the cytoplasm is positively correlated with activation of NLRP3 [95,96]. In one study, after CA-074-Me was applied to inhibit cathepsin B, the level of NLRP3 activation decreased and a similar decrease was seen in the cathepsin B level [97]. This experiment indicated that cathepsin B is an upstream signal for NLRP3 and that inhibition of it suppresses NLRP3 activation. However, studies have found that in cathepsin-deficient macrophages, NLRP3 inflammasomes can be activated by particulate matter, which indicates that cathepsin B is one of the signals that activate NLRP3, but not the only one (Figure 2) [98,99].
NLRP3-MEDIATED PYROPTOSIS:
When cells are subjected to harmful stimuli or under stress, NLRP3 activation accompanies the exposure of the N-terminal thermoprotein region, which can bind to ASC [100,101]. Another terminus of ASC contains the recruitment domain CARD, which recruits caspase-1 precursors to assemble the mature NLRP3 inflammasomes [102,103]. Procaspase-1 is cleaved by the NLRP3 inflammasome to become an activated form of caspase-1 [104]. The latter cleaves the pro-GSDMD and the pro-inflammatory factors IL-18 and IL-1β, transforming them into a mature state. Mature GSDMD has pore-forming properties, forming cavities in the cell membrane to cause cell cytoplasmic exudation and changes in osmotic pressure. IL-18 and IL-1β flow out of the cytoplasm from the pores made by the GSDMD, leading to sterile inflammation [105].
MORPHOLOGIC ALTERATIONS OF PYROPTOSIS:
Both pyroptosis and apoptosis are programmed cell death, but there are considerable differences between them. Morphologically, apoptotic cells have chromatin condensation (pyknosis), DNA fragmentation, plasma membrane blebbing, and they shed apoptotic bodies [106]. Because apoptosis is an innate cellular mechanism of programmed suicide, it does not cause cytosolic contents to be released into the extracellular environment, which would not lead to an inflammatory response. Pyroptotic cell changes are characterized by cellular swelling and formation of numerous bubble-like protrusions visible under light microscopy [107]. When pyroptotic cell walls begin to swell, chromatin condensation and DNA fragmentation is visible on electron microscopy [108]. With the formation of pores in a pyroptotic cell, the cell ruptures and the outflow of its intracellular contents is accompanied by inflammation.
NLRP3-MEDIATED PYROPTOSIS AND MI/R INJURY:
Early positive revascularization therapy has been shown to vastly decrease the size of AMI-associated MIs and to improve prognosis. However, persistent reperfusion therapy can induce an extensive range of inflammatory damage, affecting up to 50% of the area of an infarct. In the acute phase following reperfusion, the myocardial tissue exposed to sublethal ischemic insults (the infarct area) and the tissue exposed to lethal damage are areas beside the necrotic tissue which are at risk. Activation of the NLRP3 inflammasome in damaged cardiomyocytes is associated with cell death and inflammatory injury, further aggravating myocardial injury [109]. Studies have demonstrated that pyroptosis is significantly associated with MI/R injury [110–112]. Pyroptosis can lead to overwhelming production of inflammatory mediators and cardiomyocyte death. Our previous research has demonstrated that NLRP3 inflammasome-mediated caspase-1 signaling pathway-induced pyroptosis plays a vital role in MI/R injury [113]. Our previous study confirmed that uric acid worsens MI/R damage in the NLRP3 inflammasome-mediated pyroptosis signaling pathway by stimulating ROS generation. Inhibiting ROS production and activation of calpain can alleviate the MI/R injury by inhibiting NLRP3 inflammasome-mediated pyroptosis. Huairui et al found that GSDMD deficiency in cardiomyocytes significantly reduced myocardial infarct size. GSDMD gene deletion also blocked H/R-induced cardiomyocyte pyroptosis and IL-18 release [114].
Studies have shown that myocardial damage in diabetic rats is more considerable than in normal rats treated with the same surgery, and it is accompanied by a higher level of pyroptosis [12,115]. This finding indicates that NLRP3 inflammasomes are activated during MI/R, which is demonstrated by NLRP3 activation and release of inflammatory factors mediated by downstream caspase-1. In vitro, the application of NLRP3 inflammasome inhibitors and caspase-1 inhibitors significantly reduced the occurrence of cell inflammatory damage and pyroptosis [116,117]. Another study showed that silencing the NLRP3 gene can inhibit formation of the NLRP3 inflammasomes and limit MI size [118,119]. Therefore, NLRP3 and caspase-1 may be potential future targets for reducing MI/R injury.
NLRP3-MEDIATED PYROPTOSIS AND VENTRICULAR REMODELING:
Immediate reperfusion therapy reduces mortality in the acute phase of AMI and ventricular remodeling and mortality in the later phase. However, rehospitalization and mortality rates in the terminal stage were higher in patients with previous AMI who had no HF [120]. Massive cardiomyocyte death leads to permanent loss of function in cardiomyocytes in the infarcted area and causes compensated hypertrophy of cardiomyocytes in surrounding regions that typically maintain cardiac output, which is known as ventricular remodeling [121]. Ventricular remodeling caused by MI is one of the most common clinical causes of terminal HF [122,123]. Cardiac ventricular remodeling, which involves exaggerated inflammatory responses and neurohumoral mechanisms, is the compensatory repair process for local and global cardiac structure and function [124,125].
Researchers have dynamically detected changes in mouse cardiac function and death rates and found that NLRP3-deficient animals had higher survival rates and better heart function [126]. The authors have pointed out that the early nociceptive/inflammatory phase is the most critical window for the effect of NLRP3 on poor myocardial healing, adverse structural remodeling, left ventricular dysfunction, and dilation. After MI/R, mice were treated with an NLRP3 inflammasome inhibitor for 2 successive weeks. One month later, the level of myocardial fibrosis and the NLRP3-mediated pyroptotic signal were visibly reduced in the mice treated with MCC950 [127]. MCC950 also increased the ejection fraction and decreased the left ventricular end-systolic volume (LVESV) in the mice that underwent a surgical procedure for MI/R. Silencing P2X7 inhibited the activation of caspase-1 in AMI and alleviated ventricular remodeling on the 7th day, which was characterized by slight ventricular dilatation and dysfunction [128]. That finding indicates that NLRP3 inflammasome-mediated pyroptosis participates in ventricular remodeling after AMI. When NLRP3 is inhibited, the rate of HF caused by ventricular remodeling is further reduced.
Beyond NLRP3, inflammatory cytokines such as tumor necrosis factor (TNF)-α [129], IL-1 [130], IL-6 [131], and C-reactive protein (CRP) [132] clearly are increased in ventricular remodeling. These inflammatory cytokines are considered to be associated with regulation of ventricular remodeling, cardiac hypertrophy, cardiac fibrosis, and decreased cardiac contractility. Left ventricular end-diastolic volume and LVESV, measured by transthoracic echocardiography, were significantly reduced after IL-1β was inhibited. Left ventricular ejection fraction and left ventricular fractional shortening (LVFS) also were increased [133]. These findings indicate that NLRP3 inflammasome-mediated pyroptosis may be an important target of pharmacological treatment for ventricular remodeling.
Treatment for NLRP3-Mediated Pyroptosis
IMPACT ON MI/R INJURY AND VENTRICULAR REMODELING:
Even though the specific molecular mechanisms involved need to be explored in more depth, the critical role of NLRP3-mediated pyroptosis in MI/IR injury and subsequent ventricular remodeling has been widely confirmed. However, in multiple studies, inhibition of NLRP3 has been shown to reduce myocardial damage and inhibit ventricular remodeling after ischemia [134–136]. To assess the effect of inhibiting NLRP3 on cardiac function during MI/R, Marchetti et al established experimental MI/R and permanent ischemia mouse models using coronary artery occlusion [137]. NLRP3 inflammasome inhibitors were administered to the mice at reperfusion. Treatment with an NLRP3 inflammasome inhibitor significantly reduced infarct size and troponin I serum levels in mouse 24 h after creation of MI/R. After 7 days, the NLRP3 inflammasome inhibitor significantly alleviated the left ventricular systolic dysfunction (LVSD) in the MI/R mice. A significant increase in left ventricular end-diastolic dimension and LVESD evidenced by left ventricle dilatation, and a decrease in LVFS evidenced by systolic dysfunction, were observed in a mouse with permanent ischemia after 7 days. The NLRP3 inflammasome inhibitor obviously reversed the changes described after MI/R. Similar studies have proven the protective effect of an NLRP3 inflammasome inhibitor on cardiac function [134]. Following is a summary of information on some inhibitors of NLRP3-mediated pyroptosis.
NLRP3 Inhibitors
COLCHICINE:
Colchicine, a tricyclic alkaloid, was developed initially for the treatment of gout. The drug was not regarded as an NLRP3 inhibitor until 2009, when it was found to inhibit the assembly and activation of NLRP3. Microtubules, essential components for intracytoplasmic localization and assembly of the NLRP3 inflammasome, are a subcellular channel for transporting NLRP3, procaspase-1, and ASC [138,139]. Colchicine can irreversibly combine with microtubule protein to inhibit microtubule formation and elongation and promote microtubule depolymerization [140]. Studies have shown that administration of colchicine can block the subsequent inflammatory response mediated by NLRP3 in monocytes that secrete IL-18 and IL-1β and positively correlate with inflammasome activation [134]. In a mouse model of AMI, treatment with oral colchicine (0.1 mg/kg/d for 7 consecutive days) significantly reduced activation of the NLRP3 inflammasome and expression of downstream pyroptosis-related proteins of NLRP3 after 24 h. After 7 days of treatment, oral colchicine dramatically reduced the scar area in the infarct zone and ventricular remodeling of the left ventricular infarction area and improved the survival rate [134]. In another mouse MI/R model, coronary artery ligation was performed for 45 min and then 400 μg/kg of colchicine was administered intraperitoneally 25 min before reperfusion. After 24 h, colchicine significantly reduced the area of MI, while levels of troponin T and verification markers were significantly reduced [141]. After 10 weeks, the degree of myocardial fibrosis in the mice in the colchicine group also was significantly reduced. These results indicate that colchicine inhibits the inflammatory response and ventricular remodeling after AMI by inhibiting NLRP3-mediated pyroptosis. Given the progress with colchicine in basic research, some clinical studies have been done showing that the drug decreases the infarct area and myocardial damage [142,143].
GLIBENCLAMIDE AND 16673-34-0:
Glibenclamide, a sulfonylurea hypoglycemic agent, lowers blood sugar mainly by inhibiting hepatic gluconeogenesis. In macrophages incubated with glibenclamide after being treated with NLRP3 stimuli, the drug was found to inhibit activation of the NLRP3 inflammasome and release of IL-1β in a dose-dependent manner [144]. The role of glibenclamide in inhibition of NLRP3-mediated pyroptosis has been proven in vivo and in vitro, but the doses of glibenclamide in vitro must be 100-fold higher than those used for clinical treatment of diabetes, which inevitably causes fatal hypoglycemia [144]. To eliminate that adverse effect, an intermediate of glibenclamide, 16673-34-0, has been designed to inhibit the NLRP3 inflammasome [135]. Zymosan A-induced peritonitis in mice, which is entirely dependent on assembly and activation of the NLRP3 inflammasome, is mediated by the pyroptotic signaling pathway [135]. The action of 16673-34-0 in alleviating the severity of peritonitis and inhibiting activation and formation of the NLRP3 inflammasome is dose-dependent. In a study of 16673-34-0, mice were subjected to 30-min myocardial ischemia followed by 24-h reperfusion to establish an MI/R model. They were given 16673-34-0 30 min before ischemia was induced and immediately on reperfusion, and then it was administered 3 times more over a 6-h period [135]. The 16673-34-0 significantly limited the expansion of infarcts and downregulated the troponin T level and expression of the NLRP3 inflammasome. These data suggest that 16673-34-0 alleviates MI/R injury by inhibiting NLRP3 inflammasome-mediated pyroptosis [135].
MCC950:
MCC950 reversibly inhibits the NLRP3 inflammasome by changing the conformation of NLRP3, inhibiting activation of the NLRP3 inflammasome. NLRP3 undergoes a structural rearrangement during the process of activation. MCC950 can non-covalently bind to the Walker B motif of the region in the vicinity of the Walker B motif to convert NLRP3 to an inactive and closed conformation, which inhibits activation of the NLRP3 inflammasome [145]. In one study, female Landrace pigs were treated with 60-min balloon occlusion of the left anterior descending artery followed by 7 days of reperfusion to establish a MI/R model. MCC950 was found to decrease the infarct size and the level of IL-18 and IL-1β when it was injected intravenous 30 min before reperfusion into the pigs that had undergone MI/R [136].
Caspase-1 Inhibitors
VX-765:
VX-765 is a highly selective inhibitor of caspase-1 function that acts by covalently modifying cysteine residues. In one study, rats were subjected to 1-h myocardial ischemia to imitate MI, followed by 2-h reperfusion to establish a MI/R model after separate i.v. injection of VX765, cangrelor, and VX765 combined with cangrelor. The combination of antiplatelet drugs and VX765 provided an extra protective effect in the rats, reducing the MI/R injury [146]. However, because the inhibitory effects were reversible, no further exploration was planned about the protective effect of VX765 in rats with MI/R injury. The experiments were repeated by Jonathan, with extension of reperfusion over 3 days. VX765 was found to decrease the infarct size in a time-dependent manner, and with the myocardial damage alleviated, caspase-1 and the downstream inflammatory factor were downregulated. VX765 has been studied in combination with antiplatelet drugs [147].
α1-ANTITRYPSIN:
α1-antitrypsin (A1AT) is an acute-phase glycoprotein secreted and synthesized by hepatocytes that increased in acute inflammation-related diseases [148,149]. A1AT downregulates the production of IL-1β, chemokine, and interferon after being added to human monocytes [150]. An increase in A1AT levels in the plasma of patients with AMI suggests that A1AT could protect against myocardial damage. In one study, mice with myocardial ischemia were treated with an intraperitoneal injection of 60 mg/kg of A1AT at reperfusion. A1AT significantly minimized infarct size and LVSD during the initial 24 h and 7 days after reperfusion. A1AT also downregulated the expression of caspase-1 but did not influence the degree of leukocyte infiltration near the infarct region [151]. A1AT inhibited activation of caspase-1 and ventricular remodeling after myocardial infarction in mice that had undergone permanent ligation of the left anterior descending coronary artery.
IL-1 INHIBITOR:
Anakinra, an exogenous recombinant human IL-1 receptor antagonist (IL-1ra), has been used to treat rheumatoid arthritis and cryopyrin-associated periodic syndrome by interfering with IL-1β binding to IL-1 reporter I [152,153]. In vivo, anakinra alleviates myocardial remodeling and left ventricular dysfunction in mice that have been subject to acute MI and reperfusion. IL-1ra inhibits IL-1 by interacting with IL-1, competing with the IL-1 reporter I and protecting the heart from inflammatory injury [154,155]. In rats with MI, overexpression of IL-1ra has been found to diminish infarct size by 50% [156]. In recent clinical experiments, anakinra has been used to treat patients with non-ST-segment-elevation acute coronary syndrome, resulting in fewer hospitalizations for HF and less worsening of the condition than in the placebo group [157]. Even though a reduction in hsCRP values was observed in the MRC-ILA Heart clinical trial, there was late recurrence of ischemic events in patients who had received anakinra once daily for 14 days [158]. There was no clear explanation for those events, but they may be associated with a rebound inflammatory mechanism. A similar phenomenon has not been observed in another clinical experiment. However, the safety and efficacy of anakinra treatment in patients with myocardial infarction needs to be further explored in clinical experiments with a large sample size.
Clinical Application and Perspectives
Reperfusion is recognized as the most effective treatment for MI. However, myocardial injury and ventricular remodeling after reperfusion are still urgent clinical problems that need to be resolved. The NLRP3 inflammasome is a form of inflammatory cell death closely related to the occurrence and development of cardiovascular disease. Exploring the activation and mechanism of NLRP3 under ischemic conditions is crucial to understanding the pathophysiological mechanism of MI/R injury and myocardial remodeling. It also provides new directions for exploring and researching new pathological mechanisms and treatment strategies for MI/R injury and ventricular remodeling after ischemia. We have described the NLRP3-mediated pyroptosis-related pathway inflammation inhibitors that have been widely studied and used in clinical practice. The clinical trial data on targeting of inhibition of NLRP3-mediated pyroptosis as a way to reduce MI/R injury and myocardial remodeling provide a theoretical and practical basis for further extensive research and clinical practice. In theory, NLRP3 inflammasome inhibitors should have pharmacological characteristics, such as fast onset, long duration of therapeutic effect, reasonable specificity, and high safety to reduce short-term and long-term damage caused by acute MI. In a previous large-scale phase III multicenter clinical study, canakinumab significantly reduced acute inflammatory response and risk of coronary restenosis in patients with AMI [159]. Therefore, we need more research to discover more specific, safe, and effective NLRP3 inflammasome inhibitors, as well as large-scale clinical trials to corroborate their clinical safety and effectiveness.
Conclusions
NLRP3 has been identified as essential in relieving MI/R injury and easing post-MI ventricular remodeling that are activated by specific stimuli. Many inhibitors of NLRP3-mediated signaling pyroptosis have been shown to vastly reduce post-MI myocardium injury, providing additional targets for alleviating MI/R injury and ventricular remodeling. Whether inhibition of NLRP3-mediated pyroptosis is associated with other adverse effects, however, requires further exploration.
Figures
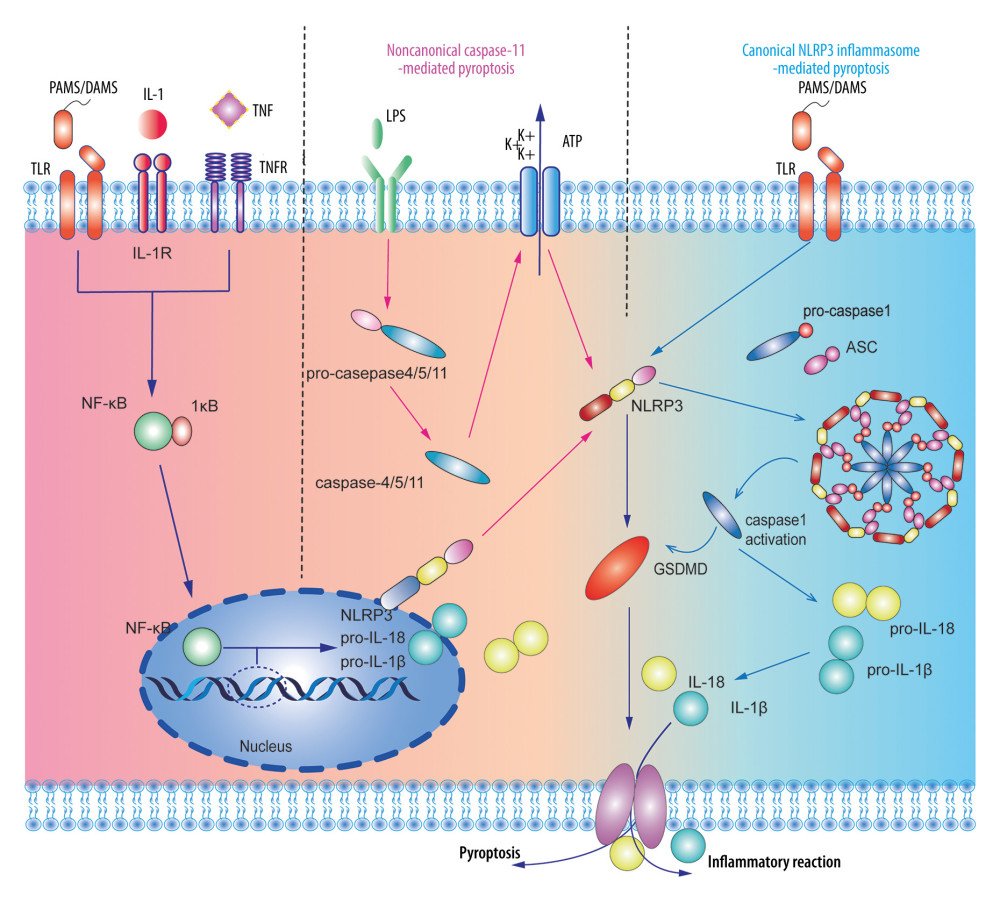 Figure 1. A dual-signal model mediated the activation process of the nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome. The first step was stimulating promotion of nuclear factor-κB into the nucleus to regulate the transcription and translation of the NLRP gene in the nucleus and release it into the cytoplasm. The second step was divided into the non-canonical caspase-11-mediated and canonical NLRP3 inflammasome-mediated pyroptosis pathways. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing.
Figure 1. A dual-signal model mediated the activation process of the nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome. The first step was stimulating promotion of nuclear factor-κB into the nucleus to regulate the transcription and translation of the NLRP gene in the nucleus and release it into the cytoplasm. The second step was divided into the non-canonical caspase-11-mediated and canonical NLRP3 inflammasome-mediated pyroptosis pathways. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing. 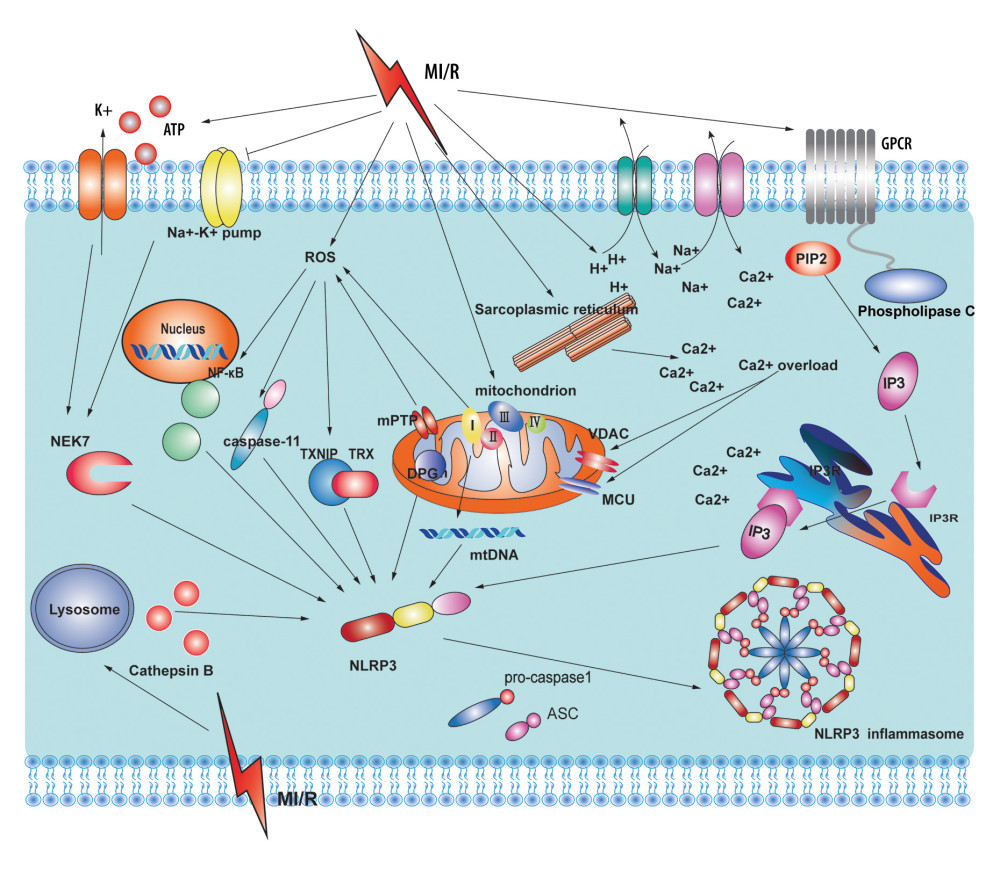 Figure 2. Potential sources and consequences of nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) activation signaling pathway during myocardial ischemia and reperfusion (MI/R). MI/R promotes potassium efflux, which directly increases the expression of NIMA-related kinase 7 (NEK7) to form the NLRP3 complex. MI/R promotes sodium-hydron exchange and sodium-calcium exchange and sarcoplasmic reticulum and endoplasmic reticulum release calcium. Calcium overload causes mitochondrial instability and NLRP3 inflammasome activation. MI/R promotes mitochondrial dysfunction and reactive oxygen species (ROS) production. ROS activates the NLRP3 inflammasomes by regulating nuclear factor-κB, caspase-11, and thioredoxin-interacting protein. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing.
Figure 2. Potential sources and consequences of nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) activation signaling pathway during myocardial ischemia and reperfusion (MI/R). MI/R promotes potassium efflux, which directly increases the expression of NIMA-related kinase 7 (NEK7) to form the NLRP3 complex. MI/R promotes sodium-hydron exchange and sodium-calcium exchange and sarcoplasmic reticulum and endoplasmic reticulum release calcium. Calcium overload causes mitochondrial instability and NLRP3 inflammasome activation. MI/R promotes mitochondrial dysfunction and reactive oxygen species (ROS) production. ROS activates the NLRP3 inflammasomes by regulating nuclear factor-κB, caspase-11, and thioredoxin-interacting protein. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing. References
1. Khan MA, Hashim MJ, Mustafa H, Global epidemiology of ischemic heart disease: Results from the global burden of disease study: Cureus, 2020; 12(7); e9349
2. Versluis A, Bank AJ, Douglas WH, Fatigue and plaque rupture in myocardial infarction: J Biomech, 2006; 39(2); 339-47
3. Kones R, Recent advances in the management of chronic stable angina I: Approach to the patient, diagnosis, pathophysiology, risk stratification, and gender disparities: Vasc Health Risk Manag, 2010; 6; 635-56
4. Heusch G, Myocardial ischemia: Lack of coronary blood flow or myocardial oxygen supply/demand imbalance?: Circ Res, 2016; 119(2); 194-96
5. Amani H, Habibey R, Hajmiresmail S, Antioxidant nanomaterials in advanced diagnoses and treatments of ischemia-reperfusion injuries: J Mater Chem B, 2017; 5(48); 9452-76
6. Jennings RB, Historical perspective on the pathology of myocardial ischemia/reperfusion injury: Circ Res, 2013; 113(4); 428-38
7. Pattar SS, Fatehi HA, Fedak PW, Acellular extracellular matrix bioscaffolds for cardiac repair and regeneration: Front Cell Dev Biol, 2019; 7; 63
8. Torrealba N, Aranguiz P, Alonso C, Mitochondria in structural and functional cardiac remodeling: Adv Exp Med Biol, 2017; 982; 277-306
9. Ali AS, Rybicki BA, Alam M, Clinical predictors of heart failure in patients with first acute myocardial infarction: Am Heart J, 1999; 138(6); 1133-39
10. Ito H, Maruyama A, Iwakura K, Clinical implications of the ‘no-reflow phenomenon: A predictor of complications and left ventricular remodeling in reperfused anterior wall myocardial infarction: Circulation, 1996; 93(2); 223-28
11. Zeng C, Duan F, Hu J, NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy: Redox Biol, 2020; 34; 101523
12. Qiu Z, Lei S, Zhao B, NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats: Oxid Med Cell Longev, 2017; 2017; 9743280
13. Wang X, Li X, Liu S, PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia: Basic Res Cardiol, 2020; 115(6); 1-14
14. Zhang J, Huang L, Shi X, Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway: Aging, 2020; 12(23); 24270-87
15. Xia X, Wang X, Zheng Y, What role does pyroptosis play in microbial infection?: J Cell Physiol, 2019; 234(6); 7885-92
16. Kesavardhana S, Malireddi RS, Kanneganti TD, Caspases in cell death, inflammation, and pyroptosis: Annu Rev Immunol, 2020; 38; 567-95
17. Zhao C, Zhao W, NLRP3 inflammasome – a key player in antiviral responses: Front Immunol, 2020; 11; 211
18. Mangan MS, Olhava EJ, Roush WR, Targeting the NLRP3 inflammasome in inflammatory diseases: Nat Rev Drug Discov, 2018; 17(8); 588-606
19. Chen IY, Moriyama M, Chang MF, Ichinohe T, Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome: Front Microbiol, 2019; 10; 50
20. O’Brien WT, Pham L, Symons GF, The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target: J Neuroinflamm, 2020; 17(1); 1-12
21. Ding S, Liu D, Wang L, Inhibiting microRNA-29a protects myocardial ischemia-reperfusion injury by targeting SIRT1 and suppressing oxidative stress and NLRP3-mediated pyroptosis pathway: J Pharmacol Exp Ther, 2020; 372(1); 128-35
22. Askarian F, Wagner T, Johannessen M, Nizet V: FEMS Microbiol Rev, 2018; 42(5); 656-71
23. Lupfer C, Kanneganti TD, The expanding role of NLR s in antiviral immunity: Immunol Rev, 2013; 255(1); 13-24
24. Proell M, Riedl SJ, Fritz JH, The NOD-like receptor (NLR) family: A tale of similarities and differences: PLoS One, 2008; 3(4); e2119
25. Bruder-Nascimento T, Ferreira NS, Zanotto CZ, NLRP3 inflammasome mediates aldosterone-induced vascular damage: Circulation, 2016; 134(23); 1866-80
26. Ketut-Carneiro N, Fitzgerald KA, Inflammasomes: Curr Biol, 2020; 30(12); R689-94
27. Elliott EI, Sutterwala FS, Initiation and perpetuation of NLRP 3 inflammasome activation and assembly: Immunol Rev, 2015; 265(1); 35-52
28. Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation: Nature, 2011; 469(7329); 221-25
29. Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling: Nat Rev Cardiol, 2014; 11(5); 255-65
30. Yi YS, Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses: Immunology, 2017; 152(2); 207-17
31. Franceschini A, Capece M, Chiozzi P, The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein: FASEB J, 2015; 29(6); 2450-61
32. Noma A, ATP-regulated K+ channels in cardiac muscle: Nature, 1983; 305(5930); 147-48
33. Rodrigo GC, The Na+-dependence of Na+-activated K+-channels (IK (Na)) in guinea pig ventricular myocytes, is different in excised inside/out patches and cell-attached patches: Pflügers Archiv, 1993; 422(5); 530-32
34. Carmeliet E, Cardiac ionic currents and acute ischemia: From channels to arrhythmias: Physiol Rev, 1999; 79(3); 917-1017
35. Mitani A, Shattock M, Physiology C, Role of Na-activated K channel, Na-K-Cl cotransport, and Na-K pump in [K] e changes during ischemia in rat heart: Am J Physiol Heart Circ Physiol, 1992; 263(2); H333-40
36. Amano F, Akamatsu Y, A lipopolysaccharide (LPS)-resistant mutant isolated from a macrophage-like cell line, J774. 1, exhibits an altered activated-macrophage phenotype in response to LPS: Infect Immun, 1991; 59(6); 2166-74
37. Yang Y, Wang H, Kouadir M, Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors: Cell Death Dis, 2019; 10(2); 1-11
38. Gong T, Yang Y, Jin T, Orchestration of NLRP3 inflammasome activation by ion fluxes: Trends Immunol, 2018; 39(5); 393-406
39. Muñoz-Planillo R, Kuffa P, K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter: Immunity, 2013; 38(6); 1142-53
40. Sanman LE, Qian Y, Eisele NA, Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death: Elife, 2016; 5; e13663
41. Jämsen E, Pajarinen J, Kouri VP, Tumor necrosis factor primes and metal particles activate the NLRP3 inflammasome in human primary macrophages: Acta Biomater, 2020; 108; 347-57
42. Shi H, Wang Y, Li X, NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component: Nat Immunol, 2016; 17(3); 250-58
43. Sharif H, Wang L, Wang WL, Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome: Nature, 2019; 570(7761); 338-43
44. Liu H, Gu C, Liu M, NEK7 mediated assembly and activation of NLRP3 inflammasome downstream of potassium efflux in ventilator-induced lung injury: Biochem Pharmacol, 2020; 177; 113998
45. Tang T, Lang X, Xu C, CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation: Nat Commun, 2017; 8(1); 1-12
46. Murphy E, Steenbergen C, Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury: Physiol Rev, 2008; 88(2); 581-609
47. Mozaffari MS, Liu JY, Abebe W, Baban B, Mechanisms of load dependency of myocardial ischemia-reperfusion injury: Am J Cardiovasc Dis, 2013; 3; 180
48. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP, The complement membrane attack complex triggers intracellular calcium fluxes leading to NLRP3 inflammasome activation: J Cell Sci, 2013; 126(13); 2903-13
49. Rüdiger S, Jung P, Shuai JW, Termination of calcium release for clustered IP3R channels: PLoS Comput Biol, 2012; 8(5); e1002485
50. Kankanamge D, Ubeysinghe S, Tennakoon M, Dissociation of the G protein βγ from the Gq-PLCβ complex partially attenuates PIP2 hydrolysis: J Biol Chem, 2021; 296; 100702
51. Chen CY, Yang CH, Tsai YF, Ugonin U stimulates NLRP3 inflammasome activation and enhances inflammasome-mediated pathogen clearance: Redox Biol, 2017; 11; 263-74
52. Colombini M, VDAC: The channel at the interface between mitochondria and the cytosol: Mol Cell Biochem, 2004; 256(1); 107-15
53. Montero M, Alonso MT, Albillos A, Mitochondrial calcium-induced calcium release mediated by the calcium uniporter: Mol Biol Cell, 2001; 12(1); 63-71
54. Murakami T, Ockinger J, Yu J, Critical role for calcium mobilization in activation of the NLRP3 inflammasome: Proc Natl Acad Sci USA, 2012; 109(28); 11282-87
55. Pizzuto M, Pelegrin P, Cardiolipin in immune signaling and cell death: Trends Cell Biol, 2020; 30(11); 892-903
56. Nakahira K, Haspel JA, Rathinam VA, Autophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA release: Nat Immunol, 2011; 12(3); 222
57. Zhong Z, Liang S, Sanchez-Lopez E, New mitochondrial DNA synthesis enables NLRP3 inflammasome activation: Nature, 2018; 560(7717); 198-203
58. Chen H, Yang D, Han F, The bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the calcium-dependent MAPK-Jnk pathway: Cell Host Microbe, 2017; 21(1); 47-58
59. Gong T, Wang X, Yang Y, Plant lectins activate the NLRP3 inflammasome to promote inflammatory disorders: J Immunol, 2017; 198(5); 2082-92
60. Abais JM, Xia M, Zhang Y, Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector?: Antioxid Redox Sign, 2015; 22(13); 1111-29
61. Becker LB, Vanden Hoek TL, Generation of superoxide in cardiomyocytes during ischemia before reperfusion: Am J Physiol, 1999; 277(6); H2240-46
62. Bugger H, Pfeil KM, Mitochondrial ROS in myocardial ischemia-reperfusion and remodeling: BBA-Mol Basis Dis, 2020; 1866(7); 165768
63. Zorov DB, Juhaszova M, Sollott SJ, Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release: Physiol Rev, 2014; 94(3); 909-50
64. Andrienko T, Pasdois P, Rossbach A, Halestrap AP, Real-time fluorescence measurements of ROS and [calcium] in ischemic/reperfused rat hearts: Detectable increases occur only after mitochondrial pore opening and are attenuated by ischemic preconditioning: PLoS One, 2016; 11(12); e0167300
65. Braunersreuther V, Montecucco F, Ashri M, Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury: J Mol Cell Cardiol, 2013; 64; 99-107
66. Matsushima S, Kuroda J, Ago T, Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α: Circ Res, 2013; 112(8); 1135-49
67. Martinon F, Signaling by ROS drives inflammasome activation: Eur J Immunol, 2010; 40(3); 616-19
68. Wu X, Zhang H, Qi W, Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis: Cell Death Dis, 2018; 9(2); 1-12
69. Saïd-Sadier N, Padilla E, Langsley G, Ojcius DM, Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase: PLoS One, 2010; 5(4); e10008
70. Hsieh TJ, Liu TZ, Lu FJ, Actinodaphnine induces apoptosis through increased nitric oxide, reactive oxygen species, and down-regulation of NF-κB signaling in human hepatoma Mahlavu cells: Food Chem Toxicol, 2006; 44(3); 344-54
71. He Y, Hara H, Núñez G, Mechanism and regulation of NLRP3 inflammasome activation: Trends Biochem Sci, 2016; 41(12); 1012-21
72. Morgan MJ, Liu Z, Crosstalk of reactive oxygen species and NF-κB signaling: Cell Res, 2011; 21(1); 103-15
73. Mohamed IN, Hafez SS, Fairaq A, Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet: Diabetologia, 2014; 57(2); 413-23
74. Zhou R, Tardivel A, Thorens B, Thioredoxin-interacting protein links oxidative stress to inflammasome activation: Nat Immunol, 2010; 11(2); 136-40
75. Han Y, Xu X, Tang C, Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis: Redox Biol, 2018; 16; 32-46
76. Mai W, Xu Y, Xu J, Berberine inhibits NOD-like receptor family pyrin domain containing 3 inflammasome activation and pyroptosis in nonalcoholic steatohepatitis via the ROS/TXNIP axis: Front Pharmacol, 2020; 11; 185
77. Cerella C, Diederich M, Ghibelli L, The dual role of calcium as messenger and stressor in cell damage, death, and survival: Int J Cell B, 2010; 2010; 546163
78. Halestrap AP, Clarke SJ, Javadov SA, Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection: Cardiovasc Res, 2004; 61(3); 372-85
79. Hernansanz-Agustín P, Enríquez JA, Generation of reactive oxygen species by mitochondria: Antioxidants-Basel, 2021; 10(3); 415
80. Rouslin W, Physiology C, Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis: Am J Physiol Heart Circ Physiol, 1983; 244(6); H743-48
81. Lesnefsky EJ, Gudz TI, Migita CT, Ischemic injury to mitochondrial electron transport in the aging heart: Damage to the iron-sulfur protein subunit of electron transport complex III: Arch Biochem Biophys, 2001; 385(1); 117-28
82. Duan J, Karmazyn M, Relationship between oxidative phosphorylation and adenine nucleotide translocase activity of two populations of cardiac mitochondria and mechanical recovery of ischemic hearts following reperfusion: Can J Physiol Pharmacol, 1989; 67(7); 704-9
83. Chiu CC, Yeh TH, Lu CS, PARK14 PLA2G6 mutants are defective in preventing rotenone-induced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway: Oncotarget, 2017; 8(45); 79046-60
84. Rated K, Rausch W-D, Gille G, Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration: Neurochem Int, 2006; 49(4); 379-86
85. Park WH, You BR, Antimycin A induces death of the human pulmonary fibroblast cells via ROS increase and GSH depletion: Int J Oncol, 2016; 48(2); 813-20
86. Byun HO, Kim HY, Lim JJ, Mitochondrial dysfunction by complex II inhibition delays overall cell cycle progression via reactive oxygen species production: J Cell Biochem, 2008; 104(5); 1747-59
87. Molagoda IMN, Lee KT, Choi YH, Anthocyanins from Hibiscus syriacus L inhibit NLRP3 inflammasome in BV2 microglia cells by alleviating NF-κB-and ER stress-induced calcium accumulation and mitochondrial ros production: Oxid Med Cell Longev, 2021; 2021; 1246491
88. Kostic M, Katoshevski T, Sekler I, Allosteric regulation of NCLX by mitochondrial membrane potential links the metabolic state and calcium signaling in mitochondria: Cell Rep, 2018; 25(12); 3465-75
89. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP, The complement membrane attack complex triggers intracellular calcium fluxes leading to NLRP3 inflammasome activation: J Cell Sci, 2013; 126(13); 2903-13
90. Shimada K, Crother TR, Karlin J, Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis: Immunity, 2012; 36(3); 401-14
91. Shokolenko I, Venediktova N, Bochkareva A, Oxidative stress induces degradation of mitochondrial DNA: Nucleic Acids Res, 2009; 37(8); 2539-48
92. Iyer SS, He Q, Janczy JR, Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation: Immunity, 2013; 39(2); 311-23
93. Acosta D, Puckett M, McMillin R, Ischemic myocardial injury in cultured heart cells: In situ lysosomal damage: Experientia, 1978; 34(10); 1388-89
94. Porter K, Lin Y, Liton PB, Cathepsin B is up-regulated and mediates extracellular matrix degradation in trabecular meshwork cells following phagocytic challenge: PLoS One, 2013; 8(7); e68668
95. Chevriaux A, Pilot T, Derangère V, Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction: Front Cell Dev Biol, 2020; 8; 167
96. Zhang Y, Chen Y, Zhang Y, Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction: Eur J Pharmacol, 2019; 865; 172795
97. Tang Y, Cao G, Min X, Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis through suppressing caspase-1 activation: J Physiol Biochem, 2018; 74(4); 503-10
98. Qi X, Man SM, Malireddi RS: J Exp Med, 2016; 213(10); 2081-97
99. Brojatsch J, Lima H, Palliser D, Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lysosome-destabilizing agents: Cell Cycle, 2015; 14(7); 964-72
100. Oroz J, Barrera-Vilarmau S, Alfonso C, ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces: J Biol Chem, 2016; 291(37); 19487-501
101. Vajjhala PR, Mirams RE, Hill JM, Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein: J Biol Chem, 2012; 287(50); 41732-43
102. Srinivasula SM, Poyet JL, Razmara M, The PYRIN-CARD protein ASC is an activating adaptor for caspase-1: J Biol Chem, 2002; 277(24); 21119-22
103. Fernandes-Alnemri T, Wu J, Yu J, The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation: Cell Death Differ, 2007; 14(9); 1590-604
104. Groß O, Yazdi AS, Thomas CJ, Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1: Immunity, 2012; 36(3); 388-400
105. Shi J, Gao W, Shao F, Pyroptosis: Gasdermin-mediated programmed necrotic cell death: Trends Biochem Sci, 2017; 42(4); 245-54
106. Böhm I, Schild H, Apoptosis: The complex scenario for a silent cell death: Mol Imaging Biol, 2003; 5(1); 2-14
107. Chen X, He W-t, Hu L, pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis: Cell Res, 2016; 26(9); 1007-20
108. Jorgensen I, Miao EA, Pyroptotic cell death defends against intracellular pathogens: Immunol Rev, 2015; 265(1); 130-42
109. Takahashi M, Cell-specific roles of NLRP3 inflammasome in myocardial infarction: J Cardiovasc Pharm, 2019; 74(3); 188-93
110. Lu C, Chen C, Chen A, Oridonin attenuates myocardial ischemia/reperfusion injury via downregulating oxidative stress and NLRP3 inflammasome pathway in mice: Evid-Based Compl Alt, 2020; 2020; 7395187
111. Wang Y, Liu X, Shi H, NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases: Clin Transl Med, 2020; 10(1); 91-106
112. Mastrocola R, Penna C, Tullio F, Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways: Oxid Med Cell Longev, 2016; 2016; 5271251
113. Shen S, He F, Cheng C, Uric acid aggravates myocardial ischemia-reperfusion injury via ROS/NLRP3 pyroptosis pathway: Biomed Pharmacother, 2021; 133; 110990
114. Shi H, Gao Y, Dong Z, GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury: Circ Res, 2021; 129(3); 383-96
115. Zhang D, He Y, Ye X, activation of autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation and attenuates myocardial ischemia-reperfusion injury in diabetic rats: J Diabetes Invest, 2020; 11(5); 1126-36
116. Leemans JC, Cassel SL, Sutterwala FS, Sensing damage by the NLRP3 inflammasome: Immunol Rev, 2011; 243(1); 152-62
117. Toldo S, Seropian IM, Mezzaroma E, Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury: J Mol Cell Cardiol, 2011; 51(2); 244-51
118. Gong Z, Pan J, Shen Q, Li M, Peng Y, Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury: J Neuroinflamm, 2018; 15(1); 1-17
119. Meng Z, Song MY, Li CF, Zhao JQ, shRNA interference of NLRP3 inflammasome alleviate ischemia reperfusion-induced myocardial damage through autophagy activation: Biochem Bioph Res Commun, 2017; 494(3–4); 728-35
120. Ezekowitz JA, Kaul P, Bakal JA, Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction: J Am Coll Cardiol, 2009; 53(1); 13-20
121. Gajarsa JJ, Kloner RA, Left ventricular remodeling in the post-infarction heart: A review of cellular, molecular mechanisms, and therapeutic modalities: Heart Fail Rev, 2011; 16; 13-21
122. Gaballa MA, Goldman S, Ventricular remodeling in heart failure: J Card Fail, 2002; 8(6); S476-85
123. Bahia MC, Kochar A, Granger CB, Post-myocardial infarction heart failure: JACC Heart Fail, 2018; 6(3); 179-86
124. Nian M, Lee P, Khaper N, Liu P, Inflammatory cytokines and postmyocardial infarction remodeling: Cir Res, 2004; 94(12); 1543-53
125. Zarrouk-Mahjoub S, Zaghdoudi M, Amira Z, Pro-and anti-inflammatory cytokines in post-infarction left ventricular remodeling: Int J Cardiol, 2016; 221; 632-36
126. Louwe MC, Olsen MB, Kaasbøll OJ, Absence of NLRP3 inflammasome in hematopoietic cells reduces adverse remodeling after experimental myocardial infarction: JACC Basic Transl Sci, 2020; 5(12); 1210-24
127. Gao R, Shi H, Chang S, The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves ventricular remodeling in a mouse model of myocardial infarction: Int Immunopharmacol, 2019; 74; 105575
128. Mezzaroma E, Toldo S, Farkas D, The inflammasome promotes adverse ventricular remodeling following acute myocardial infarction in the mouse: Proc Natl Acad Sci USA, 2011; 108(49); 19725-30
129. Sun M, Chen M, Dawood F, Tumor necrosis factor-α mediates ventricular remodeling and ventricular dysfunction after pressure overload state: Circulation, 2007; 115(11); 1398-407
130. Razin T, Melamed-Book N, Argaman J, Interleukin-1α dependent survival of cardiac fibroblasts is associated with StAR/STARD1 expression and improved ventricular remodeling and function after myocardial infarction: J Mol Cell Cardiol, 2021; 155; 125-37
131. Huo S, Shi W, Ma H, Alleviation of inflammation and oxidative stress in pressure overload-induced ventricular remodeling and heart failure via IL-6/STAT3 inhibition by raloxifene: Oxid Med Cell Longev, 2021; 2021; 6699054
132. Świątkiewicz I, Magielski P, Kubica J, C-reactive protein as a risk marker for post-infarct heart failure over a multi-year period: Int J Mol Sci, 2021; 22(6); 3169
133. Li Z, Hu S, Huang K, Targeted anti-IL-1β platelet microparticles for cardiac detoxing and repair: Sci Adv, 2020; 6(6); eaay0589
134. Fujitsue K, Sugamura K, Kurokawa H, Colchicine improves survival, left ventricular remodeling, and chronic cardiac function after acute myocardial infarction: Circ J, 2017; 81(8); 1174-82
135. Marchetti C, Chojnacki J, Toldo S, A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury following ischemia-reperfusion in the mouse: J Cardiovasc Pharm, 2014; 63(4); 316
136. Van Hout GP, Bosch L, Ellenbroek GH, The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction: Eur Heart J, 2017; 38(11); 828-36
137. Marchetti C, Toldo S, Chojnacki J, Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and non-ischemic injury in the mouse: J Cardiovasc Pharm, 2015; 66(1); 1-8
138. Misawa T, Takahama M, Kozaki T, Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome: Nat Immunol, 2013; 14; 454-60
139. Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation: Nature, 2011; 469(7329); 221-25
140. Bhattacharyya B, Panda D, Gupta S, Banerjee M, Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin: Med Res Rev, 2008; 28(1); 155-83
141. Akodad M, Fauconnier J, Sicard P, Interest of colchicine in the treatment of acute myocardial infarct responsible for heart failure in a mouse model: Int J Cardiol, 2017; 240; 347-53
142. Deftereos S, Giannopoulos G, Angelidis C, Anti-inflammatory treatment with colchicine in acute myocardial infarction: A pilot study: Circulation, 2015; 132(15); 1395-403
143. Giannopoulos G, Angelidis C, Kouritas VK, Usefulness of colchicine to reduce perioperative myocardial damage in patients who underwent on-pump coronary artery bypass grafting: Am J Cardiol, 2015; 115(10); 1376-81
144. Lamkanfi M, Mueller JL, Vitari AC, Glyburide inhibits the cryopyrin/NALP3 inflammasome: J Cell Biol, 2009; 187(1); 61-70
145. Coll RC, Hill JR, Day CJ, MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition: Nat Chem Biol, 2019; 15(6); 556-59
146. Yang XM, Downey JM, Cohen MV, The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor: J Cardiovasc Pharm T, 2017; 22(6); 574-78
147. Audia JP, Yang XM, Crockett ES, Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y 12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function: Basic Res Cardiol, 2018; 113(5); 1-15
148. Silverman EK, Sandhaus RA, Clinical practice. Alpha1-antitrypsin deficiency: N Engl J Med, 2009; 360(26); 2749-57
149. Prachalias AA, Khalife M, Francavilla R, Liver transplantation for alpha-1-antitrypsin deficiency in children: Transpl Int, 2000; 13(3); 207-10
150. Pott GB, Chan ED, Dinarello CA, Shapiro L, α-1-Antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood: J Leukocyte Biol, 2009; 85(5); 886-95
151. Toldo S, Seropian IM, Mezzaroma E, Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury: J Mol Cell Cardiol, 2011; 51(2); 244-51
152. Edrees H, Fahmy E, Mostafa H, Protective effect of apelin, amlodipine and anakinra in ischemia-reperfusion injury in myocardium: Inter J Med, 2014; 3(1); 1-8
153. Grothusen C, Hagemann A, Assmann T, Impact of an interleukin-1 receptor antagonist and erythropoietin on experimental myocardial ischemia/reperfusion injury: Sci World J, 2012; 2012; 737585
154. Patti G, Mega S, Pasceri V, Interleukin-1 receptor antagonist levels correlate with extent of myocardial loss in patients with acute myocardial infarction: Clin Cardiol, 2005; 28(4); 193-96
155. Patti G, Di Sciascio G, D’Ambrosio A, Prognostic value of interleukin-1 receptor antagonist in patients undergoing percutaneous coronary intervention: Am J Cardiol, 2002; 89(4); 372-76
156. Suzuki K, Murtuza B, Smolenski RT, overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis: Circulation, 2001; 104(Suppl 1); 308-13
157. Abbate A, Trankle CR, Buckley LF, Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction: J Am Heart Assoc, 2020; 9(5); e014941
158. Morton AC, Rothman AM, Greenwood JP, The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study: Eur Heart J, 2015; 36(6); 377-84
159. Ridker PM, Everett BM, Thuren T, Antiinflammatory therapy with canakinumab for atherosclerotic disease: N Engl J Med, 2017; 377(12); 1119-31
Figures
Figure 1. A dual-signal model mediated the activation process of the nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome. The first step was stimulating promotion of nuclear factor-κB into the nucleus to regulate the transcription and translation of the NLRP gene in the nucleus and release it into the cytoplasm. The second step was divided into the non-canonical caspase-11-mediated and canonical NLRP3 inflammasome-mediated pyroptosis pathways. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing.Figure 2. Potential sources and consequences of nucleotide-binding and oligomerization-like receptor pyrin domain-containing protein 3 (NLRP3) activation signaling pathway during myocardial ischemia and reperfusion (MI/R). MI/R promotes potassium efflux, which directly increases the expression of NIMA-related kinase 7 (NEK7) to form the NLRP3 complex. MI/R promotes sodium-hydron exchange and sodium-calcium exchange and sarcoplasmic reticulum and endoplasmic reticulum release calcium. Calcium overload causes mitochondrial instability and NLRP3 inflammasome activation. MI/R promotes mitochondrial dysfunction and reactive oxygen species (ROS) production. ROS activates the NLRP3 inflammasomes by regulating nuclear factor-κB, caspase-11, and thioredoxin-interacting protein. Adobe Illustrator CC 2019 (Adobe Software, San Jose, California, United States) was used for drawing. In Press
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952