28 December 2021: Review Articles
A Review of the Impact of Neutrophils and Neutrophil Extracellular Traps (NETs) on the Development of Aortic Aneurysms in Animal and Human Studies
Milena MichalskaDOI: 10.12659/MSM.935134
Med Sci Monit 2021; 27:e935134






Abstract
ABSTRACT: The pathogenesis of the aortic aneurysm (AA) includes several mechanisms, such as chronic sterile inflammation and homeostasis imbalance, with arteriosclerosis, hemodynamic forces, and genetic factors. In addition to the roles of these processes in the development of AA, neutrophilic activity may play a pivotal role (mostly in inflammation and thrombus formation). Neutrophils, which play a crucial role in innate immunity, can release neutrophil extracellular traps (NETs), one of the mechanisms against fighting pathogens, beside phagocytosis and degranulation. NETs are structures composed of nuclear elements (eg, chromatin and modified histones) and granular and cytoplasmic components, which can lead to inflammation and coagulation changes. In addition, the exacerbation of NETosis (the process of NET formation) can be noticed in vascular diseases, including in the development of AA and myocardial infarction and in diabetes, hypertension, and COPD, which are the risk factors of the presence of AA. The discharge of NETs, which are extracellular materials formed by citrullinated histones (Cit-H), cell-free DNA fibers (cf-DNA), and granular and cytoplasmic molecules, is a newly identified method of neutrophil activation that can be activated by endogenous inflammatory stimuli, which contribute to AA development. Cit-H and cf-DNA can be used as biomarkers of AA growth. By understanding the neutrophilic influence of NET release, a new pathway of screening AA growth (by measurement of biomarkers of NETosis) and pharmacological assessment (by repression of NET formation) can be developed. This review summarizes the current knowledge about the influence of NETs on AA growth in human and animal studies.
Keywords: Neutrophils, Inflammation, aortic aneurysm, Animals, atherosclerosis, Endothelium, Vascular, Extracellular Traps, Humans, Thrombosis
Background
The pathophysiology of the aortic aneurysm (AA) is complex and not fully understood. Some cellular mechanisms can exist together in the abdominal aortic aneurysm (AAA) and thoracic aortic aneurysm (TAA), and some are more characteristic of one of them. The development of AA requires changes in smooth muscle cells (SMC), endothelial cells, lymphocytes, monocytes/macrophages, platelets, and the subject of this review, neutrophils [1]. Activated neutrophils can lead to dysfunction between the endothelium and neutrophils, resulting in changes in coagulation and the inflammatory system [2]. Neutrophils are immune system cells that aid in the defense of the body against invading microorganisms and they migrate as the first-line of defense to infectious tissues. They are critical for the normal functioning of the innate immune response because of their ability to respond quickly in the presence of pathogens. Neutrophils can use several mechanisms, such as phagocytosis, degranulation, and oxidative burst, to immediately kill intruders and are connected with the oxidative burst release of neutrophil extracellular traps (NETs) in the process known as NETosis [3]. The etiology of AA growing based on interdependencies between neutrophilic activity (mainly on NETosis) and injury of the aortic wall is multifactorial and includes relationships between selectins, chemokines, cytokines, and other types of vascular and bloodstream cells [4].
Use of Animal Models in AA
The use of animal models, notably of mice and rats, has been the most prevalent method of investigating the mechanisms involved in AAA pathophysiology. Conventional rodent-based models can include angiotensin II infusion (Ang II), elastase perfusion, calcium chloride (CaCl2) or calcium phosphate application, and xenograft (transplantation of aorta from 1 species to another, for example, guinea pig to rat) [5]. Table 1 lists the most commonly used models and some of their properties, compared with those of human AAA. The mechanism of developing AAA in animal models can differ depending on the methods used. SMC depletion, localized chronic medial and adventitial inflammation, elastin fiber breakdown, and medial attenuation define the modified aortic wall [6]. Several unique rodent models have been identified, some of which combine elements of the conventional models mentioned above but are modified to better reflect human AAA. For example, using the feed-forward mechanism of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis in the development of TAA in Marfan syndrome mice, which are targeted with anti-TGFβ or folic acid diet, seems to be the new pathway for understanding the development of AA [7]. Thrombosis, triglyceride-rich adipocytes within the adventitia, and regional neutrophilic activity have all been implicated in the etiology and pathophysiology of the disease in clinical investigations, although few animal models have mirrored these characteristics [8]. The most common AAA models include non-dissected aneurysms, unlike in human pathogenesis in which enlarged vascular intramural hematomas can occur from dissection, which we can observe only in Ang II perfusion [9]. The general limitations of the use of animal models were excluded because a large part of the description of pathogenesis was based on animal studies that do not 100% reflect the formation of AA in humans. The major constraint is that the development of AA in humans takes decades, whereas aortic dilatation in animal models takes days to weeks. In humans, chronic remodeling with the healing process is documented; however, in animal models, the occurrence of aneurysm is too rapid to examine this process. An additional concern is the experimentally established topography of aneurysms along the aorta. The study of the origin of aneurysms in humans determines their aortic topography, with a high predilection of the ascending aorta for degeneration and monogenic abnormalities, and a considerable proclivity for the infrarenal segment in connection with atherosclerosis. It is questionable whether the animal model’s various localizations of aneurysms along the aorta represent the same mechanism of development of AA in humans. Another limitation of animal models is that the use of triggering factors causes the initiation of aortic dilatation more than it causes the progression of the existing aneurysms. Whereas the occurrence of aneurysm in humans is a more long-term process, with a progression and stabilization stage, compared to animals in which the acute phase is characterized by a uniform transition from a healthy aorta to dilatation. All of these factors should be considered when evaluating experimental model results.
Pathogenesis of Aortic Aneurysm and Neutrophilic Role
The understanding of the pathogenesis of AA has been developing for 30 years. The process of AA includes complex relationships between the aortic wall, hemodynamic forces, blood component, and immune response. To simplify, the current etiology of AA is divided into main pathways, including inflammation, homeostasis imbalance with emphasis on arteriosclerosis (which were included in this review), hemodynamics, and genetic factors. However, AA is a result of all processes in varying degrees.
Inflammation
Many studies have strongly suggested that sterile chronic inflammation plays a critical role in the development and progression of AA. The elasticity and strength of the aortic wall are based on the extracellular matrix (ECM) components produced by SMC in the aortic media, such as laminin, gelatin, collagen, and proteoglycan [12]. The proteolytic degradation of ECM by metalloproteinase (MMP) causes SMC apoptosis and inflammatory cell infiltration [13]. MMPs include more than 20 zinc-dependent proteolytic enzymes that can also promote adhesion of clotting factors, lipoproteins, and chemotactic molecules [14]. The polymorphisms of MMP incorporate degeneration of collagenases (MMP-1, MMP-8, and MMP-13), gelatinases (MMP-2 and MMP-9), stromelysins (MMP-3 and MMP-10), and another MMP group (MMP-12) [13]. In their meta-analysis, Tan Li et al suggested that MMP-2, MMP-3, and MMP-13 are significantly correlated with the risk factors of AAA, and MMP-2 and MMP-8 are associated with thoracic aortic dissection. MMP-9 has a crucial role in increasing the risk of AAA. MMP-9 is produced mostly by neutrophils and also by endothelial cells and fibroblasts, which begin SMC migration, macrophage infiltration, and neo-angiogenesis in the medial layer, as shown by immunostaining of a human AAA specimen. Strong positive Cd4 staining signifies a neo-formed structure in the medial layer [15]. In Marfan syndrome, patients’ levels of MMP-9 and MMP-2 are significantly increased [16], and doxycycline, as a non-specific MMP inhibitor, can inhibit this secretion [17]. Mice with Marfan syndrome spontaneously die between 2 to 4 months from the rupture of the thoracic aorta [18]. Xiong et al, in a Marfan syndrome mice model, showed prolonged survival of the mutant mice that received doxycycline treatment, with reduced MMP expression and improved histopathological signs of aortic matrix degradation [17]. In a clinical trial of patients with AAA that evaluated the effect of doxycycline used 2 weeks before open repair, there was significantly reduced aortic wall MMP-3 and MMP-25 mRNA and reduced protein levels of neutrophil collagenase (MMP-8) and MMP-9. In terms of the apparent selective effect on neutrophil-associated proteases in aortic wall neutrophil content, which was validated by immunohistochemistry, a 75% reduction in aneurysm wall neutrophil content was demonstrated. These data strongly suggest that doxycycline reduced neutrophilic influx in an aneurysmal wall, showing why neutrophilic activity (due to interleukin [IL]-8 hyperexpression) is a prominent feature of AAA [19]. To test the hypothesis that neutrophils play an important role in AAA, a study of neutrophil depletion (by anti-neutrophil-antibody treatment) limited experimental AAA formation by altering MMP-2 and/or MMP-9 in mice that underwent aortic elastase perfusion, showing the importance of neutrophils in the early stages of experimental AAA development. Results revealed that the percentage of mice forming AAAs after elastase perfusion was 67% in the mice control group and 8% in the anti-neutrophil-antibody group [20]. Tissue inhibitors of metalloproteinases (TIMPs) are important enzymes for maintaining the balance between the degradation of the ECM by MMP. Decreased amounts of TIMPs can be found in aneurysmal extracts, as compared with in atherosclerotic and normal controls. Reduced TIMPs may be secondary due to the inflammatory reaction and excessive neutrophil elastase activity. TIMP is found throughout the aortic medium, mostly in an extracellular position [21]. Unlike the large group of MMPs, the TIMP group consists of only 4 members, TIMP1-4. TIMP-1 and TIMP-2 are not upregulated; however, TIMP-3 is significantly upregulated. In the presence of increased MMP activity, the absence of increased TIMP-1 or TIMP-2 reflects an unbalanced proteolytic state. Others have found that TIMP-3 expression is much higher in AAAs in the entire aneurysm tissue, while gene expression and protein synthesis are both lower in TAAs [22]. The specific involvement of TIMP-3 in AAA formation is further confirmed by a TIMP-3 genetic mutation (nt-1296) that has been linked to familial AAA [23]. Some TIMP genes are located on the X-chromosome, which is indicated by the higher incidence of AAA in men than women.
Soluble blood components, such as inflammatory cells, are transferred and accumulated in the tunica media through the highly vascularized adventitia, resulting in infiltration of inflammatory cells into the vascular media due to the destruction of the ECM structure by MMPs and loss of tunica media resistance [24]. A variety of inflammatory cells transfer through the tunica adventitia; this process is initiated by perivascular tissue, which causes infiltration of several cells, such as neutrophils, macrophages, natural killer cells, dendritic cells, B and T lymphocytes, and mast cells. All of these inflammatory cells are thought to play roles in the development of AAA, and their interactions create the inflammatory milieu of the aorta walls [25]. The inhibition of inflammatory cells by toll-like receptors in CaCl2 animal models [26] and in humans showed significant changes in the development of AAA [27]. Chronic inflammation is linked to increased vascular reactive oxygen species (ROS) production, which includes a group of chemicals generated from oxygen, such as nitric oxide and superoxide. Intracellular ROS can be generated by a variety of enzymes, including xanthine oxidase, NOS, and the NADPH oxidases, which can activate MMP (MMP-9) [28]. NADPH oxidases and inducible NOS (iNOS) have a causal role because reduced ROS generation in the aortic tissues is caused by an iNOS deficiency or suppression of NADPH oxidase and is directly linked to the prevention of matrix degradation and aneurysm development in CaCl2-treated wild-type mice [28]. ROS not only activate MMP but also cause vascular cell injury and lipid peroxidation, which contribute to vascular remodeling and explain the mechanism by which ROS are involved in arteriosclerosis and hypertension (by modulation of Ang II), which are the risk factors for developing AAA [29]. Neutrophils can modulate ROS by releasing myeloperoxidase, which was shown in 2 animal models (Ang II perfusion and CaCl2) [30]. AAA was reduced when myeloperoxidase was depleted, and a similar impact was seen when taurine, an amino acid with antioxidant characteristics, was given [30].
Atherosclerosis and Thrombosis
During the last few decades, the main way of understanding developing AAA has been based on atherosclerosis, but the novel pathways suggest that lipid dysregulation is the result of chronic inflammation, which seems to be more important in the etiology of this disease. This is because atherosclerotic plaque is discovered in most cases of AAA [31], implying that atherosclerosis is not a necessary factor for AAA development.
For understanding the genetic and pharmacological background, mouse models of atherosclerosis and AAA remain the best method for addressing the function of specific molecules and underlying molecular pathways. Mice without the lipoprotein transporter apolipoprotein E (ApoE−/−) and low-density lipoprotein receptor-deficient mice (LDL-R−/−) are 2 genetically modified mouse strains commonly utilized in atherosclerosis research. ApoE−/− mice have spontaneous atherosclerosis, which is aggravated when the mice are fed a high-fat diet. LDL-R−/− mice, on the other hand, develop atherosclerotic lesions only when fed a high-fat diet [32]. Endothelin has been linked to the development of human atherosclerosis in AAA. In ApoE−/− mice, endothelin overexpression is exacerbated with atherosclerosis from a high-fat diet. Endothelin-induced ROS and inflammation can have a role in the progression of atherosclerosis and the development of AAA [33]. The traditional “oxidative stress theory” of atherosclerosis is based on the production of ROS by resident cells in vessels and other organs and tissues, which causes the oxidation of LDLs, resulting in inflammatory responses and the formation of foam cells within atherosclerotic plaques [29].
In the pathogenesis of atherosclerosis, neutrophils have often been overlooked because of their minor role in atherosclerosis in humans. Much of this is owing to the uncommon detection of neutrophils in atherosclerotic lesions, which can be explained by their short lifespan and ability to undergo phenotypic changes, displaying markers typically expressed on antigen-presenting cells, thus appearing as macrophage-like or dendritic cell-like cells. Another reason that neutrophils have been overlooked is the lack of sensitive and specific methods of detection. This is exacerbated further by the use of staining methods that are not neutrophil-specific [34]. The release of prepared granule proteins, which are ejected into the surroundings of activated neutrophils, is responsible for much of the neutrophil-dependent pro-inflammatory activity. Neutrophils are the only cells that express granule proteins, such as azurocidin, α-defensins, and NGAL. Interestingly, immunohistochemistry identified all of these granule proteins in human atherosclerotic lesions, implying that these proteins could constitute neutrophil fingerprints [34]. In contrast, pure neutrophil cells are not seen in human atherosclerosis and do not have a pivotal role in the etiology of atherosclerotic AAs.
NETosis in Vascular Pathologies
GENERAL BACKGROUND OF NETOSIS:
NETosis is a mechanism by which NETs are released by neutrophils, which are made up of modified chromatin and bactericidal proteins from granules and cytoplasm. NETosis can be induced by a variety of infections, antibodies, and immunological complexes and also by cytokines, microcrystals, or physiological stressors. ROS are required for the induction of NETosis. Granule components are released into the cytosol, modified histones lead to chromatin decondensation, and the nuclear envelope is destroyed during NETosis [35]. The role of histones is fungicidal and bactericidal but can also be cytotoxic for endothelial cells; thus, tissue damage and septic death can be triggered by extracellular histones [36]. Two pathways of NETosis are currently described. The first is called suicidal or classic (sometimes called also lytic) because it was reported by Brinkmann and Zychlinsky in 2004 to cause the death of neutrophils [37]. The second, which was characterized later, is vital NETosis, in which the cell keeps its viability as well as many of its effector functions [37]. DNA, myeloperoxidase, neutrophil elastase, cathepsin G, lactoferrin, calprotectin, lactoferrin, gelatinase, proteinase-3, and peptidoglycan-binding protein are all components of NETs having inflammatory and bactericidal properties [38]. NETs have long been recognized as an important pathogen barrier, especially by binding microorganisms, inhibiting their dissemination, and maintaining a high local concentration of antimicrobial chemicals released by neutrophils, which can lead to the death of microorganisms. Also, NETs are important in non-pathogenic infections and have been linked to a variety of cardiovascular disorders and with risk factors of diseases, including diabetes, arteriosclerosis, venous thrombosis, and obesity [39].
NETOSIS AND ENDOTHELIAL REGULATION:
The endothelium, the innermost layer of cells that covers the vasculature, serves as a dynamically changeable interface that helps the cardiovascular system maintain homeostasis. Endothelial cells can stimulate the interaction with neutrophils during transmigration. The result of this process is NETosis, as activated endothelial cells are present in inflammatory circumstances. This interaction could amplify the inflammatory response [40]. It was observed that NETs can kill endothelial cells and promote thrombosis [41]. The balance between anticoagulation and immune response activities is maintained by vascular endothelial cells. Endothelial dysfunction is linked to atherosclerosis and venous thromboembolism, 2 important cardiovascular disorders [42]. Endothelial disfunction is expressed by decreased vasodilatation and a pro-inflammatory condition manifested by the increased production of adhesion molecules and chemokines, which lead to enhanced leakiness [43]. When neutrophils are activated by atherosclerosis-related stimuli, myeloperoxidase, an enzyme contained in the main granules of neutrophils, is also released in the process of NET formation. In patients with hyperlipidemia, circulating neutrophils contain decreased amounts of myeloperoxidase, although plasma myeloperoxidase levels are increased, suggesting that the neutrophils discharge the granules. Myeloperoxidase plasma levels that are elevated may not only serve as a biomarker, but they may also have significant pathophysiological aspects. Myeloperoxidase may have a role in the initiation of endothelial dysfunction by limiting nitric oxide bioavailability. Myeloperoxidase attaches to the ECM and transforms chloride anion plus hydrogen peroxide into hypochlorous acid, a powerful oxidant and chlorinating species. Myeloperoxidase-produced hypochlorous acid may also play a role in endothelial cell desquamation and the development of a prothrombotic phenotype. Physiologically appropriate amounts of hypochlorous acid were found to cause endothelial cell death and can cause superficial erosion in arteriosclerosis plaque [42].
NETS AND ATHEROSCLEROSIS:
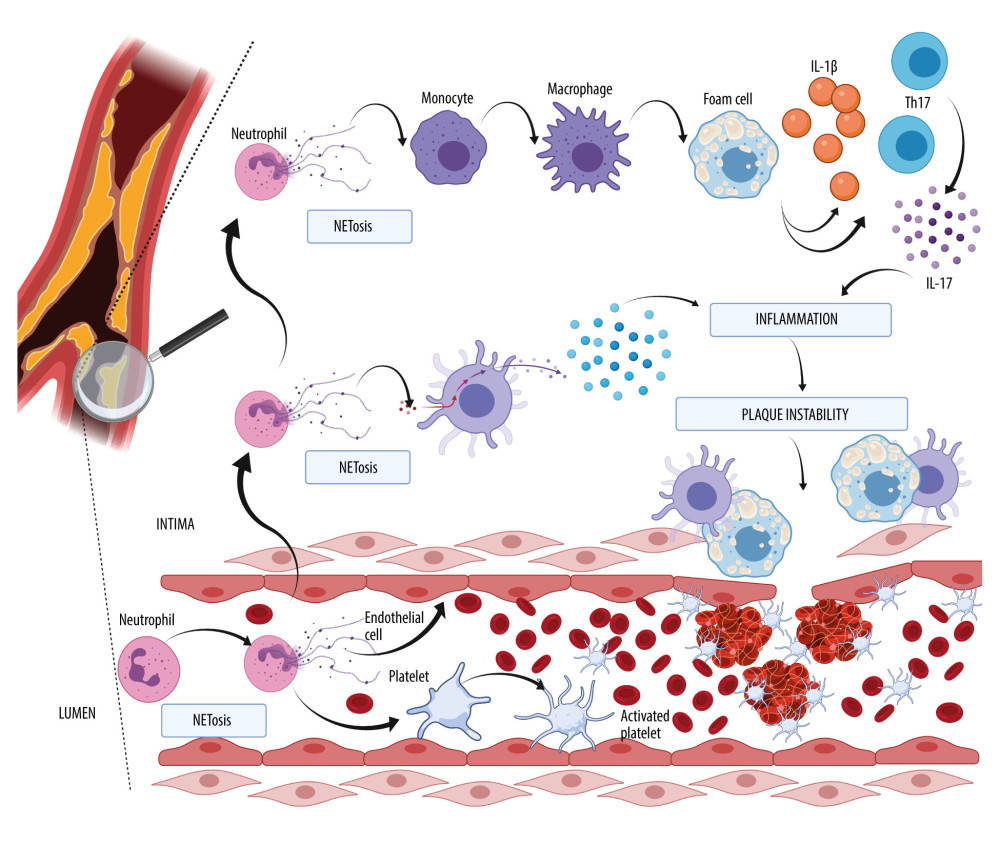
Recent studies have suggested a strong impact of NETosis on atherosclerosis development, which is commonly known as lipid-driven inflammation of the arteries. As lesions progress, it can result in plaque instability and a rupture-inducing intra-luminal thrombus, leading to ischemic events. NETs can be found in human and mouse atherosclerotic lesions [44]. Two proatherosclerotic interleukins play an essential role in atherogenesis: IL-1β and the less researched IL-18. Mice deficient in IL-1β released by macrophages develop smaller atherosclerotic lesions and less inflammation, even in the presence of hypercholesterolemia [45]. Atherosclerosis and other inflammatory pathologies are exacerbated by the presence of IL-1β. Cholesterol crystallizes in circulation because it is poorly soluble and is taken up by monocyte-derived macrophages, which activate their inflammasomes and cause them to generate IL-1β and other pro-inflammatory cytokines. These chemicals attract myeloid cells to the endothelium of blood vessels, where their cholesterol causes obstructive lesions [46]. Experimental models using murine elastase-induced perfusion revealed that IL-1β is required for the development of AAA. The amount of IL-1β protein in the aorta was much higher in murine elastase-perfused mice than in the control group, which corresponds to the recruitment of neutrophils to AAA. Both genetic IL-1 deficiency (IL-1-knockout mice) and pharmacological inhibition of IL-1 binding to the IL-1 receptor (via IL-1RA/anakinra treatment) protected mice from AAA formation, with lower aortic infiltration of neutrophils and macrophages, indicating that aortic IL-1 synthesis is required for AAA formation. These findings show that infiltrating neutrophils can be exposed to high levels of IL-1β in AAA by NETosis [47]. NETs promote macrophages into cytokine production and activate T helper 17 cells (Th17), which release IL-17 [48]. According to an experimental model using a solution of elastase perfusion, IL-17 is a key mediator of AAA formation, and CD4+ T cells are its source. Gene expression and protein levels of IL-17 were significantly elevated in aortic tissue from AAA, compared with a control group [49]. The known initiation factor is apolipoprotein B, which can accumulate in the subendothelial layer and, with NETosis, can enhance migration of blood monocytes into intima by interaction with endothelial cells, a process in which excess chemokines are involved. Cholesterol has an influence on the maturation of monocytes; thus, hypercholesterolemia increases the number of monocytes (monocytosis), and cholesterol is correlated with neutrophilia. Elevated levels of tumor necrotic factors (TNF) and IL-17 promote the production of G-CSF by bone marrow, resulting in an increased number of neutrophils [42]. Lipid-loaded macrophages transform into foam cells. The number of atherosclerotic foam cells can be increased by gene-targeting experiments in ApoE-deficient mice. [50]. The study of animals lacking apolipoprotein E allowed us to identify the ApoE-independent pathway of NET-driven atherogenesis, including dendritic cell activation by the following mechanism: dendritic cells in the vessel wall are stimulated by granule proteins and DNA complexes released in NETosis, which results in a robust type I interferon response that promotes atherogenesis. Plaque load and type I interferon response were both decreased when dendritic cells were depleted [51].
The role of NETosis in the formation of atherosclerotic plaque is shown in Figure 1. Note, NETosis can exist in the lumen and in the wall of the vessel. The atherosclerotic plaque is distinguished by infiltration by immune cells, such as monocytes, which transform into macrophages, T cells, dendritic cells, and also what is best characterized by an accumulation of lipids in foam cells. The production of a fibrous cap formed primarily of collagen by vascular smooth cells can rupture and trigger atherothrombosis by the exposure to thrombogenic particles, which is prompted by platelet aggregation, coagulation, and the formation of emboli. Platelets and neutrophils in the lumen can activate each other, resulting in NET formation and contributing to a prothrombotic milieu for thrombus development. In the intima, neutrophils and the NETs connected with them can modulate the lengthy process of atherothrombosis plaque growth by interaction with dendritic cells and the release of type I interferon. Neutrophils and NETs can play a role in the generation of foam cells from macrophages, and thus IL-1β and IL-17 released by Th17 can contribute to the growth of plaque formation, which consequently leads to instability and rupture into the lumen, where thrombus can arise.
NETS AND ATHEROTHROMBOSIS:
The development of a thrombus within a lumen in an artery that mostly results from plaque instability is known as atherothrombosis. Neutrophils and macrophages and interaction between neutrophils and platelets play significant roles in the formation of an embolus. Thrombosis is a local consequence of advanced atherosclerosis, and neutrophils are a component of the thrombus [52]. Most commonly, atherothrombosis occurs after an atheroma rupture, which can be exacerbated by NETs [53,54]. In addition to platelets, neutrophils quickly concentrate at areas of damage following plaque rupture, where they can stimulate platelet activation and initiate blood clotting. In the arterial thrombi of individuals with myocardial infarction, neutrophils dominate. Cytokines such as TNF-α and IL-1 activate neutrophils. Platelets and endothelium are attracted to activated neutrophils by adhesion molecules. P-selectin and adhesion molecules are also expressed by active platelets on their surface membrane, which mediates mechanical contact with neutrophils. Most neutrophils in arterial thrombi stain positive for citrullinated histone 3 (Cit-H3), an indicator for neutrophil priming toward NET formation. An inhibitor of peptidylarginine deaminase 4 (PAD4, which is necessary for NET formation) with Cl-amidine (hydrochloride) decreased atherosclerosis load in mice, as well as the risk of arterial thrombosis, and further minimized myocardial infarction damage [55]. Platelets and red blood cells are trapped and aggregate because NETs can act as mechanical scaffolding, and plasma proteins, such as fibrinogen, fibronectin, or von Willebrand factor, can also be caught in NETs, increasing clot formation [56]. NETs were commonly found in newly formed thrombi but not in older, more structured cardiac thrombus specimens. There was a positive correlation between infarct size and ST-segment resolution, indicating that coronary thrombus NET load can be clinically relevant in myocardial infarction [57]. As a result, it may be hypothesized that NETs are engaged in the development of coronary thrombus formation and lytic degradation.
Process of Visualization and Measurement of NETs
To determine the formation of NETs, several useful techniques are used, including in vitro and in vivo methods. Microscopic techniques like immunofluorescence microscopy, transmission electron microscopy (TEM), and scanning electron microscopy (SEM) have been successfully used in determining the presence of NETs by adequate visualization and quantification, which correspond with pathways of induction of releasing of NETs. However, in situ techniques aid with identifying the meaning of NETosis during the changing periods of diseases. TEM and SEM were used by Brinkmann et al for the initial discovery of NET release [37]. While TEM has been used to investigate the morphology of NETs, SEM is better for revealing the construction [58]. One of the most characteristic elements that is liberated is myeloperoxidase, an enzyme that produces hypochlorite anion using chlorine and hydrogen peroxide as substrates, neutrophil elastase (an enzyme which is able to collapse the outer membrane of bacteria), histones (especially citrullinated histones), cathepsin G, cathelicidins, lactoferrin, and gelatinases [3]. Several proteins, such as myeloperoxidase or neutrophil elastase, are commonly used as a marker of NETosis and are connected with other parameters, such as citrullinated histones or released cell-free DNA (cf-DNA) [59]. DNA in the nucleus of neutrophils lead to decondensation in the beginning of NETosis and, by migration of enzyme PAD4, form cytoplasm into the nucleus, where citrullination of histones take place. In this process, the most obvious markers Cit-H3 and Cit-H4 emerge, which can be examined, for example, by imaging flow cytometry [47,60]. Flow cytometry enables us to examine thousands of samples in a very short time; however, the limitation of this procedure is that Cit-H3-deficient mice can still produce some NETs, which is shown by staining with Cit-H3 antibodies [61]. Cf-DNA liberated from neutrophils are also a marker of NETosis, which can be measured by fluorescence spectroscopy using PicoGreen (Invitrogen), which is sensitive for double-stranded DNA. The disadvantage of this method is that double-stranded DNA can also be released during cell necrosis.
Resolvin D1 (RvD1) is one of the D-series resolvin mediators that decrease the inflammation exacerbated by NETosis. Resolvins belong to polyunsaturated fatty acid metabolites. RvD1 was shown to decrease AAA development by interfering with NETosis in a mouse model [62].
NETs in AAA
As shown in Table 2, a search for previous characterizations of neutrophilic influence on AA progression identified 9 studies in PubMed and Scholar Google. The search criteria included “NET”/”NETs” or “neutrophil extracellular traps” and “AAA” or “abdominal aortic aneurysm”, “TAA”, or “thoracic aortic aneurysm”. Duplications were excluded. Table 2 shows the 9 studies with the most important information.
Epidemiological studies revealed a connection between periodontal disease and the progression of AAA. The finding of bacterial DNA in tissue samples from the aorta suggested that weak bacteria can play a role in AAA progression [63]. Delbosc et al researched neutrophilic recruitment in human and animal models by repeated retention of the anaerobic pathogen
Dipeptidyl peptidase I (DPPI) is a cysteine protease member of the papin family expressed in several tissues and cells, such as mast cells, cytotoxic T cells, and myelomonocytic cells [65]. The pivotal role of DPPI is the activation of granule-associated serine proteases, such as proteinase-3, cathepsin G, and neutrophil elastase, which regulate the neutrophilic activity in the inflammation process. Mice with DPPI function loss are not susceptible to the formation of elastase-induced experimental AAAs [66]. Yan et al revealed the influence of NETs, which was independent of infection, in aortic tissue in a human and elastase-induced murine model. The markers myeloperoxidase, histones, and DNA were observed mostly in adventitia in the wild-type mice aorta, where neutrophils can easily accumulate. Significantly reduced NET formation was observed during DNase1 treatment in wild-type animals, in which heat inactivation of DNase1 eliminated this action. Furthermore, delayed DNase1 treatment after maximum neutrophil activity did not contribute to the suppression of AAA. Thus, the role of neutrophils and NETs is most important in the acute phase of an inflammatory cascade. This process was observed in animal models, in which the aorta is exposed to a single insult for developing an aneurysm, as opposed to humans, in which the process of AAA formation consists of several phases of exacerbation and stabilization. Neutrophil elastase and proteinase-3 are important for AAA development because mice with a deficiency in neutrophil elastase and NERP-3 have a smaller diameter of AA. Cathelicidins are immunomodulatory peptides with antimicrobial functions. Cathelicidin-related antimicrobial peptide (CRAMP) may be bound in NETs and present as complexes between CRAMP-DNA, which interfere with dendritic cells and INF type I, which consequently promote AAA in DPPI-deficient mice [67].
The IL-1 family consists of 11 cytokines that are involved in inflammatory responses and 10 receptors. The most representative are IL-1α and IL-1β, expressed by B lymphocytes, natural killer cells, and endothelial cells [68]. IL-1β protein and gene expression in aortic tissue and circulating in the bloodstream are elevated in patients with AAA compared with in healthy controls. A murine model with IL-1β-KO mice revealed >50% reduction in maximal aortic dilatation compared with wild-type mice. Thus, inhibition and delayed inhibition of the IL-1R pathway by anakinra prevent AAA formation in mice. It could be hypothesized that IL-1β has an influence on acute- and late-phase AAA progression. Anti-IL-1β treatment seems to be a good pharmacological target because it is safe in humans and efficiently attenuates the inflammation process [69]. Meher et al observed the dominant importance of IL-1β in NETosis in AAA development with ceramide synthesis [47]. Ceramides are lipid molecules that are in high concentrations in the cell membrane. Proapoptotic C16-ceramide is synthesized by ceramide synthase 6 [70]. In human and animal studies by Meher et al, IL-1β antibodies showed co-localization with markers of NETosis, mostly on the border of the intima and media. NETosis revealed dose-dependent induction by IL-1β; consequently, we can say that IL-1β-induced NETosis can be attended by IL-1RA. This process requires ceramide synthesis, as confirmed by a significant increase of ceramide 16 in AAA samples. To check the IL-1β expression by neutrophils, wild-type and IL-1-KO mice underwent elastase infusion. The neutrophil concentration in the blood was similar in both groups, but aortic infiltrated neutrophils were significantly lower in IL-1β-KO mice. To reduce murine AAA formation, 2 inhibitors were used: Cl-amidine (inhibitor of PAD4) and DNase. DNase did not affect aortic tissue, but Cl-amidine attenuated AAA formation, probably because PAD4 inhibitors stop the beginning of the process, while DNase interrupts the final result of NETosis [47].
Resolvins are bioactive metabolomes that can limit innate immunological responses and antimicrobial activity. The name “resolvin” refers to the distinct structures and temporal codes involved in production during resolution [71]. Because the resolvins can alleviate inflammation, in contrast to NETosis, it seems reasonable to use resolvins to attenuate AAA progression. Spinosa et al used an animal model (Ang II- and elastase-induced) to check RvD1 interaction. The results revelated attenuated AAA formation with RvD1 treatment in the elastase-induced model (day 14) and a decreased amount of Cit-H3 (day 3), while in controls it was much higher. In the second model (Ang II perfusion), RvD1 alleviated AAA formation and decreased neutrophilic indicators (Cit-H3). IL-10 was increased in the RvD1-treated group, while IL-1β was alleviated. It can be concluded that IL-1β and IL-10 work in a contrary fashion in AAA formation: IL-10 decreases inflammation, while IL-1β increases inflammation. RvD1 also works on pathway other than inhibiting NETosis because it decreases MMP-2 concentration [62]. These findings imply that RvD1 might be used as a medicinal treatment to prevent AAA development.
Visona et al looked at immunophenotypic analysis of tissue from acute medial dissection of the aorta and examined the immunohistochemistry to conventional histology of the sequential 4 phases of injury (acute, subacute, early organizing, and scarring) [72]. The first phase, acute phase (I), includes only fresh blood cells, with hardly any granulocytes. The second phase, subacute infiltrating (II) phase, is characterized by a huge number of granulocytes. In the third phase, early organizing (III), smooth muscle ingrowth and vessels in hematomas are seen. In the fourth and last phase, scarring (IV), collagen scars with few inflammatory cells are noticed. The concentration of neutrophils was evaluated in connection with the production of NETs within hematomas and the surrounding artery wall. Acute dissections included a large amount of myeloperoxidase released by neutrophils, while Cit-H3 was rare or nonexistent. The number of neutrophils and NETs rose considerably in the subacute phase II group but fell markedly in the stage III and IV groups. Curiously, neutrophils and NETs were found in the periadventitial fat tissues as well, with the highest numbers in stage II, followed by a substantial reduction in stages III and IV. Macrophages did not show a strong time correlation with dissection age but they were detected inside of hematomas in the intima and media layer. The study by Visona et al confirms the complexity of an inflammatory cascade in AA, with neutrophils as the first migration cells, then macrophages and T lymphocytes. Transmigration of neutrophils and macrophages from the vasa vasorum in phases I and II causes an increased amount of these cells in the adventitia, which can be important in arterial response to injury. In contrast to previous studies, this study showed that neutrophilic activity is noticed not only in early phases but also later [72].
The degradation process of NETs is poorly understood. Haider et al proved that macrophages could degrade NETs through extracellular predigestion by DNase fragmentation (mainly by DNase 1L3), followed by absorption [73]. In addition, their study indicated that pro-inflammatory macrophages promote NET breakdown by increasing macropinocytosis, preparing them for NET engulfment. The researchers postulated that macrophages can destroy NETs in vivo and inhibit macropinocytosis (by imipramine) in mice with a higher NET level in thrombi, consequently leading to decreased thrombi resolution. In AAA samples in which expanded macrophages were noticed, the presence of NETs was decreased. The study was performed in a human and animal model (murine vena cava thrombosis model) and confirmed that NETs can form in a human blood clot. Macrophages are recruited monocytes that transmigrate through the vessel walls and, based on inflammatory conditions, may represent a pro-inflammatory or anti-inflammatory response. Pro-inflammatory polarization enhances macropinocytosis, which stimulates NET absorption by macrophages [73].
The main message from the research of Eilenberg et al is that Cit-H3 can be a novel indicator for AAA progression based on animal (Ang II- and elastase-induced) and human models [74]. Even antiplatelet and hypolipidemic treatment did not affect Cit-H3 levels in patients with AAA. The prognostic value is higher than the diagnostic value because NETosis markers can be elevated in other cardiovascular diseases, such as hypertension and myocardial infarction. The results showed that myeloperoxidase and Cit-H3 were highly correlated with cf-DNA-histone complexes and Cit-H3 is weakly matched with a neutrophilic count. This suggests that NET concentration is strongly based on neutrophilic activity rather than on the total amount of neutrophils. It was shown that a cut-off level of Cit-H3 in plasma was determined to be 194 ng/mL based on Youden’s index, indicating fast development (≥2 mm/6 months) with 77% sensitivity and 64% specificity. Accordingly, patients with AAA in which the Cit-H3 plasma level was higher than the median had an odds ratio of 3.94 (95% confidence interval, 1.15–13.58) for fast progression, whereas those in the 4th quartile compared with the 1st quartile had an odds ratio of 8.25 (95% confidence interval, 1.15–59.00). To assess the real value of Cit-H3 as a prognostic marker of AAA progression, the test group with AAA needs to be much larger. In the Ang II-induced murine model, the citrullination blockage of histones attenuated the progression of AAA, which was associated with decreased citrullinated histones. GSK484 (selective PAD4 inhibitor) was used to stop the NETosis in both murine models (with Ang II and elastase), but in the elastase-induced representation, the PAD4 inhibitor failed to block the progression of AAA. The differences may have come from dissimilar AAA pathogenesis in both models, while usage of elastase is characterized more by acute inflammatory response with a sudden influx of mediators, and in Ang II representation, we can notice aortic dissection [74].
In the next study, Wei et al investigated how PAD4 inhibition affected NETosis in an Ang II-induced AAA murine model [75]. YM3-56 was used as a PAD inhibitor (similar to Cl-amidine but with better bioavailability and increased permeability), and the blockage of this signal causes a reduced aortic diameter, decreases mortality in AAA mice, and alleviates apoptosis of SMC through the p38/JNK pathway. YM3-56 had an influence on the rupture of AAA but not on the initiation of an aneurysm. The results confirmed that Ang II-induced NET formation in vivo and in vitro, and therefore increased NETosis markers were observed. YM3-56 decreased the concentration of Cit-H3 and double-stranded DNA, and attenuated MMP-2 expression, which alleviates degradation of ECM, thereby increasing elastin fibers degradation. Apoptosis of vascular SMC was caused by the transfer of information by the p38/JNK pathway which is induced by NETosis [75]. The p38/JNK pathway is part of the MAPK signaling pathway, which leads to Jun or p53 phosphorylation and the induction of FasL links to Fas phosphorylation. Thus, increasing the ratio between Bax and Bcl-2 activates cytochrome c and caspase cascade [76].
The last study refers to essential hypertension, in which Ang II triggers NET release. AAA specimens were collected from only 3 patients with AAA and essential hypertension; therefore, the study group was very small, and the study has therefore been briefly described here. In untreated patients newly diagnosed with essential hypertension, tissue factor concentration and markers of NETosis were increased. Patients treated with Ang II-receptor blockers revealed increased NET components and thrombin activity. NET residues have been found in AAA tissue slices, mostly in the subendothelial region of the damaged AAA wall, near the destroyed elastic lamina [77]. The activation of the NET/TF/thrombin axis exacerbates the procoagulant condition of essential hypertension, increasing vascular injury [77].
NETs in Other Diseases that Can Be Risk Factors of AAA
NETs are commonly described in cardiovascular, immunological, and metabolic diseases. This indicates that neutrophil activity can independently play a role as a risk factor in the pathophysiological mechanism of disease. As described previously, since NETosis plays a role in thrombosis, it seems reasonable to investigate this process in coronary artery diseases. The concentrations of neutrophilic markers, like ds-DNA and nucleosomes, are correlated with the coronary aortic burden with ST resolution and infarct size [78]. Neutrophils can infiltrate the pancreas with diabetes mellitus types 1 and 2, and the localization of ds-DNA and myeloperoxidase shows that NETosis can exist in pancreatic conditions. Neutrophil elastase and proteinase-3 increases in patients with diabetes mellitus type 1 are correlated with NETosis [79,80]. The role of NETs in diabetes mellitus type 2 is a little controversial, although an increased glucose level induces NETosis in vitro and in vivo [81]. Obesity is classified as an inflamed condition because fatty tissue failure impairs adipocytokine synthesis. Inflammatory cells can also invade fatty tissue and produce pro-inflammatory compounds. In preclinical and clinical investigations, NETs were found to have an importance in obesity-related inflammation [79,81]. Neutrophilic inflammation is thought to play a role in the development of chronic obstructive pulmonary disease. Although the bactericidal role is useful against an invasion of pathogens, NETs can cause considerable harm to the host. Although NETs have been linked to the pathophysiology of chronic lung disease, their involvement in chronic obstructive pulmonary disease is under investigation [79,82]. NETs may induce epithelial and endothelial apoptosis, compromising pulmonary function and hastening disease development [83].
Conclusions
The activity of neutrophils in AA development was overlooked and underestimated by researchers for several years of exploring this process. Since NETosis was described in 2004, the role of neutrophils in the pathophysiology of vascular diseases has received more interest. The relevance of neutrophil activity in the development of AA is multifocal, mostly at the beginning of sterile inflammation, which has an essential role in the pathogenesis of AAA. ROS can cause vascular injury, lipid peroxidation, and remodeling of the vascular wall. Neutrophils can regulate this process by releasing myeloperoxidase and other granularity. NETosis can play an essential role in atherosclerosis and the development of thrombi in the aortic lumen through an interface between endothelial cells. NETs probably play a key role in the development of AAA, as shown by several studies, which were mostly done in animal models. The outcome of studies showed that NETs are abundant in the intra-luminal thrombus of AAA, and there are increased concentrations of neutrophilic markers in aortic specimens and blood plasma/serum. To confirm this theory, more human studies need to be done, including evaluation of NETs before the aortic diagnosis, during the growth, and after the surgical approach. Understanding the role of NETosis in the development of AAA can impact the discovery of new therapeutic options and introduce into practice novel biomarkers of AAA enlargement.
References
1. Quintana RA, Taylor WR, Cellular mechanisms of aortic aneurysm formation: Circ Res, 2019; 124(4); 607-18
2. Meegan JE, Yang X, Coleman DC, Neutrophil-mediated vascular barrier injury: Role of neutrophil extracellular traps: Microcirculation, 2017; 24(3); e12352
3. Papayannopoulos V, Zychlinsky A, NETs: A new strategy for using old weapons: Trends Immunol, 2009; 30(11); 513-21
4. Plana E, Oto J, Medina P, Novel contributions of neutrophils in the pathogenesis of abdominal aortic aneurysm, the role of neutrophil extracellular traps: A systematic review: Thromb Res, 2020; 194; 200-8
5. Etienne H, Journé C, Rouchaud A, Persistence of intraluminal thrombus makes saccular aneurysm more biologically active than fusiform in an experimental rat model: J Vasc Res, 2020; 57(3); 164-76
6. Shimizu K, Mitchell RN, Libby P, Inflammation and cellular immune responses in abdominal aortic aneurysms: Arterioscler Thromb Vasc Biol, 2006; 26(5); 987-94
7. Huang K, Wang Y, Siu KL, Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics: Redox Biol, 2021; 38; 101757
8. Tanaka H, Zaima N, Sasaki T, Imaging mass spectrometry reveals a unique distribution of triglycerides in the abdominal aortic aneurysmal wall: J Vasc Res, 2015; 52(2); 127-35
9. Tanaka H, Zaima N, Kugo H, The role of animal models in elucidating the etiology and pathology of abdominal aortic aneurysms: Development of a novel rupture mechanism model: Ann Vasc Surg, 2020; 63; 382-90
10. Lysgaard Poulsen J, Stubbe J, Lindholt JS, Animal models used to explore abdominal aortic aneurysms: A systematic review: Eur J Vasc Endovasc Surg, 2016; 52(4); 487-99
11. Patelis N, Moris D, Schizas D, Animal models in the research of abdominal aortic aneurysms development: Physiol Res, 2017; 66(6); 899-915
12. Henderson EL, Geng Y-J, Sukhova GK, Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms: Circulation, 1999; 99(1); 96-104
13. Li T, Lv Z, Jing J-J, Yang J, Yuan Y, Matrix metalloproteinase family polymorphisms and the risk of aortic aneurysmal diseases: A systematic review and meta-analysis: Clin Genet, 2017; 93(1); 15-32
14. Brinckerhoff CE, Matrisian LM, Matrix metalloproteinases: A tail of a frog that became a prince: Nat Rev Mol Cell Biol, 2002; 3(3); 207-14
15. Ramella M, Boccafoschi F, Bellofatto K, Endothelial MMP-9 drives the inflammatory response in abdominal aortic aneurysm (AAA): Am J Transl Res, 2017; 9(12); 5485-95
16. Nataatmadja M, Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm: Circulation, 2003; 108(90101); 329II-34
17. Xiong W, Knispel RA, Dietz HC, Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome: J Vasc Surg, 2008; 47(1); 166-72
18. Pereira L, Lee SY, Gayraud B, Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1: Proc Natl Acad Sci USA, 1999; 96(7); 3819-23
19. Abdul-Hussien H, Hanemaaijer R, Doxycycline therapy for abdominal aneurysm: Improved proteolytic balance through reduced neutrophil content: J Vasc Surg, 2009; 49(3); 741-49
20. Eliason JL, Hannawa KK, Ailawadi G, Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation: Circulation, 2005; 112(2); 232-40
21. Brophy CM, Marks WH, Reilly JM, David Tilson M, Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: A preliminary report: J Surg Res, 1991; 50(6); 653-57
22. Morris DR, Biros E, Cronin O, Kuivaniemi H, Golledge J, The association of genetic variants of matrix metalloproteinases with abdominal aortic aneurysm: A systematic review and meta-analysis: Heart, 2013; 100(4); 295-302
23. Airhart ND, Brownstein B, Schierding W, Smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin: J Vasc Surg, 2013; 57(5); 23S
24. Yuan Z, Lu Y, Wei J, Abdominal aortic aneurysm: Roles of inflammatory cells: Front Immunol, 2021; 11; 609161
25. Guerriero JL, Macrophages: Their untold story in T cell activation and function: Int Rev Cell Mol Biol, 2019; 342; 73-93
26. Yan H, Cui B, Zhang X, Antagonism of toll-like receptor 2 attenuates the formation and progression of abdominal aortic aneurysm: Acta Pharm Sin B, 2015; 5(3); 176-87
27. Jabłońska A, Neumayer C, Bolliger M, Analysis of host toll-like receptor 3 and RIG-I-like receptor gene expression in patients with abdominal aortic aneurysm: J Vasc Surg, 2018; 68(6); 39-46S
28. Xiong W, Mactaggart J, Knispel R, Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model: Atherosclerosis, 2009; 202(1); 128-34
29. Chen Q, Wang Q, Zhu J, Reactive oxygen species: Key regulators in vascular health and diseases: Br J Pharmacol, 2018; 175(8); 1279-92
30. Kim HW, Blomkalns AL, Ogbi M, Role of myeloperoxidase in abdominal aortic aneurysm formation: mitigation by taurine: Am J Physiol Heart Circ Physiol, 2017; 313(6); H1168-79
31. Madaric J, Vulev I, Bartunek J, Frequency of abdominal aortic aneurysm in patients >60 years of age with coronary artery disease: Am J Cardiol, 2005; 96(9); 1214-16
32. Peshkova IO, Schaefer G, Koltsova EK, Atherosclerosis and aortic aneurysm – is inflammation a common denominator?: FEBS J, 2016; 283(9); 1636-52
33. Li MW, Mian MOR, Barhoumi T, Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein e knockout mice: Arterioscler Thromb Vasc Biol, 2013; 33(10); 2306-15
34. Soehnlein O, Multiple roles for neutrophils in atherosclerosis: Circ Res, 2012; 110(6); 875-88
35. Vorobjeva NV, Chernyak BV, NETosis: Molecular mechanisms, role in physiology and pathology: Biochemistry (Moscow), 2020; 85(10); 1178-90
36. Muñoz LE, Kaplan MJ, Radic M, Herrmann M, Editorial: NETosis 2: The excitement continues: Front Immunol, 2017; 8; 1318
37. Brinkmann V, Reichard U, Goosmann C, Neutrophil extracellular traps kill bacteria: Science, 2004; 303(5663); 1532-35
38. Bonaventura A, Liberale L, Carbone F, The pathophysiological role of neutrophil extracellular traps in inflammatory diseases: J Thromb Haemost, 2018; 118(1); 6-27
39. Mozzini C, Pagani M, Cardiovascular diseases: Consider netosis: Curr Probl Cardiol, 2021 [Online ahead of print]
40. Gupta AK, Joshi MB, Philippova M, Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death: FEBS Lett, 2010; 584(14); 3193-97
41. Villanueva E, Yalavarthi S, Berthier CC, Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus: J Immunol, 2011; 187(1); 538-52
42. Qi H, Yang S, Zhang L, Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis: Front Immunol, 2017; 8; 928
43. Pieterse E, Rother N, Garsen M, Neutrophil extracellular traps drive endothelial-to-mesenchymal transition: Arterioscler Thromb Vasc Biol, 2017; 37(7); 1371-79
44. Megens RTA, Vijayan S, Lievens D, Presence of luminal neutrophil extracellular traps in atherosclerosis: Thromb Haemost, 2012; 107(03); 597-98
45. Hansson GK, Hermansson A, The immune system in atherosclerosis: Nat Immunol, 2011; 12(3); 204-12
46. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V, Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis: Science, 2015; 349(6245); 316-20
47. Meher AK, Spinosa M, Davis JP, A novel role of IL-1β in NETosis and abdominal aortic aneurysms: Arterioscler Thromb Vasc Biol, 2018; 38(4); 843-53
48. Pejnovic N, Vratimos A, Lee SH, Increased atherosclerotic lesions and Th17 in interleukin-18 deficient apolipoprotein E-knockout mice fed high-fat diet: Mol Immunol, 2009; 47(1); 37-45
49. Sharma AK, Lu G, Jester A, Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment: Circulation, 2012; 126(11 Suppl 1); S38-45
50. Moore Kathryn J, Tabas I, Macrophages in the pathogenesis of atherosclerosis: Cell, 2011; 145(3); 341-55
51. Döring Y, Manthey HD, Drechsler M, Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis: Circulation, 2012; 125(13); 1673-83
52. Döring Y, Soehnlein O, Weber C, Neutrophil extracellular traps in atherosclerosis and atherothrombosis: Circ Res, 2017; 120(4); 736-43
53. Pertiwi KR, de Boer OJ, Mackaaij C, Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis: J Pathol, 2019; 247(4); 505-12
54. Pircher J, Engelmann B, Massberg S, Schulz C, Platelet-neutrophil crosstalk in atherothrombosis: Thromb Haemost, 2019; 119(08); 1274-82
55. Knight JS, Luo W, O’Dell AA, Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis: Circ Res, 2014; 114(6); 947-56
56. Fuchs TA, Brill A, Duerschmied D, Extracellular DNA traps promote thrombosis: Proc Nat Acad Sci USA, 2010; 107(36); 15880-85
57. Mangold A, Alias S, Scherz T, Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size: Circ Res, 2015; 116(7); 1182-92
58. de Buhr N, von Köckritz-Blickwede M, How neutrophil extracellular traps become visible: J Immunol Res, 2016; 2016; 4604713
59. Gavillet M, Martinod K, Renella R, Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples: Am J Hematol, 2015; 90(12); 1155-58
60. Barbu EA, Dominical VM, Mendelsohn L, Thein SL, Detection and quantification of histone H4 citrullination in early NETosis with image flow cytometry version 4: Front Immunol, 2020; 11; 1335
61. Li P, Li M, Lindberg MR, Kennett MJ, PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps: J Exp Med, 2010; 207(9); 1853-62
62. Spinosa M, Su G, Salmon MD, Resolvin D1 decreases abdominal aortic aneurysm formation by inhibiting NETosis in a mouse model: J Vasc Surg, 2018; 68; 93-103S
63. Aoyama N, Suzuki J, Wang D, Porphyromonas gingivalis promotes murine abdominal aortic aneurysms via matrix metalloproteinase-2 induction: J Periodont Res, 2010; 46(2); 176-83
64. Delbosc S, Alsac J-M, Journe C: PLoS One, 2011; 6(4); e18679
65. Wolters PJ, Laig-Webster M, Caughey GH, Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways: Am J Respir Cell Mol Biol, 2000; 22(2); 183-90
66. Pagano MB, Bartoli MA, Ennis TL, Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms: Proc Nat Acad Sci, 2007; 104(8); 2855-60
67. Yan H, Zhou H-F, Akk A, Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation: Arterioscler Thromb Vasc Biol, 2016; 36(8); 1660-69
68. Mantovani A, Dinarello CA, Molgora M, Garlanda C, IL-1 and related cytokines in innate and adaptive immunity in health and disease: Immunity, 2019; 50(4); 778-95
69. Johnston WF, Salmon M, Su G, Genetic and pharmacologic disruption of interleukin-1β signaling inhibits experimental aortic aneurysm formation: Arterioscler Thromb Vasc Biol, 2013; 33(2); 294-304
70. Lu P, White-Gilbertson S, Nganga R, Expression of the SNAI2 transcriptional repressor is regulated by C16-ceramide: Cancer Biol Ther, 2019; 20(6); 922-30
71. Serhan CN, Levy BD, Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators: J Clin Investig, 2018; 128(7); 2657-69
72. Visonà SD, de Boer OJ, Mackaaij C, Immunophenotypic analysis of the chronological events of tissue repair in aortic medial dissections: Cardiovasc Pathol, 2018; 34; 9-14
73. Haider P, Kral-Pointner JB, Mayer J, Neutrophil extracellular trap degradation by differently polarized macrophage subsets: Arterioscler Thromb Vasc Biol, 2020; 40(9); 2265-78
74. Eilenberg W, Zagrapan B, Bleichert S, Histone citrullination as a novel biomarker and target to inhibit progression of abdominal aortic aneurysms: Transl Res, 2021; 233; 32-46
75. Wei M, Wang X, Song Y, Inhibition of peptidyl arginine deiminase 4-dependent neutrophil extracellular trap formation reduces angiotensin II-induced abdominal aortic aneurysm rupture in mice: Front Cardiovasc Med, 2021; 8; 676612
76. Kuo W-H, Chen J-H, Lin H-H, Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway: Chem Biol Interact, 2005; 155(1–2); 31-42
77. Chrysanthopoulou A, Gkaliagkousi E, Lazaridis A, Angiotensin II triggers release of neutrophil extracellular traps, linking thromboinflammation with essential hypertension: JCI Insight, 2021; 6(18); e148668
78. Helseth R, Solheim S, Arnesen H, The time course of markers of neutrophil extracellular traps in patients undergoing revascularisation for acute myocardial infarction or stable angina pectoris: Mediat Inflamm, 2016; 2016; 182358
79. Klopf J, Brostjan C, Eilenberg W, Neumayer C, Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease: Int J Mol Sci, 2021; 22(2); 559
80. Valle A, Giamporcaro GM, Scavini M, Reduction of circulating neutrophils precedes and accompanies type 1 diabetes: Diabetes, 2013; 62(6); 2072-77
81. Bonaventura A, Vecchié A, Abbate A, Montecucco F, Neutrophil extracellular traps and cardiovascular diseases: An update: Cells, 2020; 9(1); 231
82. Pullan J, Greenwood H, Walton GM, Neutrophil extracellular traps (NETs) in COPD: A potential novel mechanism for host damage in acute exacerbations: Eur Respir J, 2015; 46; PA5055
83. Trivedi A, Khan MA, Bade G, Talwar A, Orchestration of neutrophil extracellular traps (Nets), a unique innate immune function during chronic obstructive pulmonary disease (COPD) development: Biomedicines, 2021; 9(1); 53
Tables
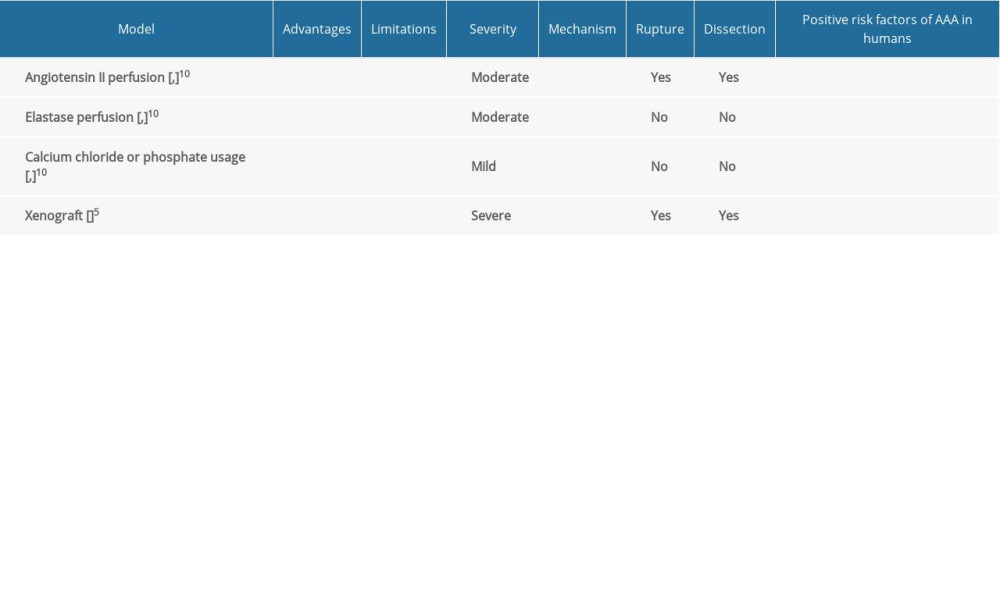 Table 1. Comparison of animal models of aortic aneurysm.
Table 1. Comparison of animal models of aortic aneurysm.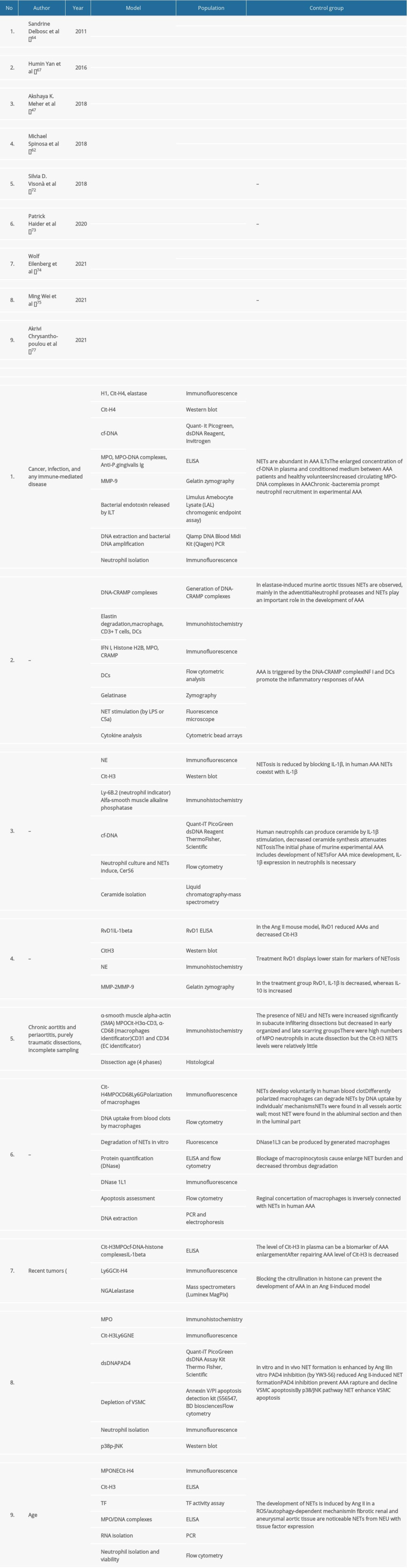 Table 2. Summary of published studies that include influence on neutrophil extracellular traps in aortic aneurysm.Table 1. Comparison of animal models of aortic aneurysm.Table 2. Summary of published studies that include influence on neutrophil extracellular traps in aortic aneurysm.
Table 2. Summary of published studies that include influence on neutrophil extracellular traps in aortic aneurysm.Table 1. Comparison of animal models of aortic aneurysm.Table 2. Summary of published studies that include influence on neutrophil extracellular traps in aortic aneurysm. In Press
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
08 Mar 2024 : Laboratory Research
Evaluation of Retentive Strength of 50 Endodontically-Treated Single-Rooted Mandibular Second Premolars Res...Med Sci Monit In Press; DOI: 10.12659/MSM.944110
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952