26 February 2022: Review Articles
A Review of Current Clinical Concepts in the Pathophysiology, Etiology, Diagnosis, and Management of Hypercalcemia
Carolina Rodrigues Tonon1ABCDEF*, Taline Alisson Artemis Lazzarin Silva1ABCDEF, Filipe Welson Leal Pereira1ABDE, Diego Aparecido Rios Queiroz1ABDE, Edson Luiz Favero Junior1BD, Danilo MartinsDOI: 10.12659/MSM.935821
Med Sci Monit 2022; 28:e935821






Abstract
ABSTRACT: Calcium is the most abundant extracellular cation in the body, and it is responsible for structural and enzymatic functions. Calcium homeostasis is regulated by 3 factors: calcitonin, vitamin D, and parathyroid hormone (PTH). Hypercalcemia is defined by a serum calcium concentration >10.5 mg/dL, and it is classified into mild, moderate, and severe, depending on calcium values. Most cases are caused by primary hyperparathyroidism and malignancies. Various mechanisms are involved in the pathophysiology of hypercalcemia, such as excessive PTH production, production of parathyroid hormone-related protein (PTHrp), bone metastasis, extrarenal activation of vitamin D, and ectopic PTH secretion. The initial approach is similar in most cases, but a definitive treatment depends on etiology, that is why etiological investigation is mandatory in all cases. The majority of patients are asymptomatic and diagnosed during routine exams; only a small percentage of patients present with severe manifestations which can affect neurological, muscular, gastrointestinal, renal, and cardiovascular systems. Clinical manifestations are related to calcium levels, with higher values leading to more pronounced symptoms. Critically ill patients should receive treatment as soon as diagnosis is made. Initial treatment involves vigorous intravenous hydration and drugs to reduce bone resorption such as bisphosphonates and, more recently, denosumab, in refractory cases; also, corticosteroids and calcitonin can be used in specific cases. This review aims to provide a clinical update on current concepts of the pathophysiology of calcium homeostasis, epidemiology, screening, clinical presentation, diagnosis, and management of hypercalcemia.
Keywords: Hyperparathyroidism, Hypercalcemia, Humoral Hypercalcemia Of Malignancy, Diphosphonates, Diagnostic Techniques, Digestive System, Disease Management, Early Diagnosis, Humans
Background
Calcium is the most abundant extracellular cation in the human body; 99% is found as a component of bones and teeth, maintaining their structure [1]. Only 1% of total calcium is circulating in the blood, 40% of which is bound to proteins, 10% is bound to organic anions, and 50% is ionized, which is the active form. Normal serum ionized calcium concentration are 1.1–1.4 mmol/L (4.5–5.6 mg/dL) [1]. Intracellular calcium gradient is maintained by Ca2+/H+ ATPases and Na+/Ca2+ exchangers [2].
In addition to maintaining bone structure, calcium acts as a second messenger; it disseminates messages to the intracellular system in response to stimuli from surface cell receptors [3]. A significant characteristic of calcium is that due to its high availability in cells, it can be quickly mobilized through calcium channels, generating a fast response [3]. Calcium acts as a cofactor for enzymes such as adenosine triphosphatase, collagenases, neutral protease, transglutaminase, and others. It also participates in the coagulation cascade, activating platelets and coagulation factors including Factor XIII [4], regulates muscle contraction, binding to troponin C on actin filaments, or calmodulin, which modulates myosin filaments [5], and stabilizes cell membranes by decreasing permeability to ions and altering membrane potential [6–8].
In situations in which serum calcium concentration increases above the normal range (calcium concentration higher than 10.5 mg/dL), we observe a condition known as hypercalcemia. This condition has several causes, and we should always investigate its etiology. Despite having different etiologies, the clinical presentation of hypercalcemia is similar in several diseases.
This review aims to provide a clinical update on current concepts of the pathophysiology of calcium homeostasis, epidemiology, screening, clinical presentation, diagnosis, and management of hypercalcemia.
Calcium Homeostasis
Corporal calcium homeostasis is regulated by 3 factors: calcitonin, vitamin D, and parathyroid hormone (PTH), which is the most important regulator [9,10]. PTH is produced by the parathyroid glands, located in close proximity to the thyroid gland lobes [9,10]. Calcium level is the main determinant for PTH production and release, which exerts negative feedback on the parathyroids [11]. Decreased serum calcium concentration stimulates PTH release, which acts mainly in bones and renal tubules; it may also interfere in intestinal calcium absorption [11]. In the bones, PTH increases the activity and number of osteoclasts, thus increasing bone reabsorption of calcium and phosphorus. In the kidneys, it stimulates tubular calcium reabsorption and phosphorus excretion and increases renal hydroxylation of 25-hydroxyvitamin D (25[OH]D) to 1,25-dihydroxyvitamin D (1,25[OH]2D) [11]. The consequence of this well-regulated mechanism is re-establishment of adequate calcium concentration [12].
Vitamin D, another important calcium regulator, is mostly produced by the skin; only a small amount comes from diet (vitamin D2 and D3) [11]. In the skin, vitamin D3 is synthesized after exposure to ultraviolet B radiation by enzyme 7-dehydrocholesterol. Vitamin D2 and D3 undergo hydroxylation in the liver by 25-hydroxylase, generating 25[OH]D, the main circulating form. In the kidneys, 1-α-hydroxylase converts 25[OH]D into 1,25[OH]2D. This form of vitamin D increases intestinal calcium absorption, which may occur passively via the paracellular route or actively [11].
The third mechanism regulating calcium homeostasis is calcitonin. Calcitonin is a hormone produced by thyroid parafollicular C cells, which is released into the circulation in response to an acute increase in calcium concentration [11]. It decreases bone and tubular calcium reabsorption. However, the role of calcitonin in calcium regulation is not yet well established; for example, after thyroidectomy, in which C cells are removed, there is no calcium disturbance [11]. The regulatory mechanisms of calcium homeostasis are summarized in Figure 1.
Several clinical conditions can disrupt the regulatory mechanisms, such as some types of cancer, medications, granulomatous diseases, chronic kidney disease, and other conditions that will be cited throughout this article, with different pathophysiologic mechanisms.
Definition, Epidemiology, and Screening for Hypercalcemia
Hypercalcemia is defined as calcium concentration higher than 10.5 mg/dL. It is classified into mild, moderate, and severe, depending on calcium values: mild 10.5–12 mg/dL, moderate 12.1–14 mg/dL, and severe >14 mg/dL [1,2,11].
Routinely, serum calcium concentration is evaluated by measuring total calcium. However, in situations such as thrombocytosis, hyperproteinemia, hypoproteinemia, multiple myeloma, or macroglobulinemia, ionic calcium should be assessed, although the test for this is not widely available [13]. When total calcium is measured, the value should be corrected by serum albumin concentration, as albumin is the main protein that carries the ion. The correction is made by the following formula: Corrected calcium (mg/dL)=Ca measured (mg/dL)+[0.8×(4–albumin (g/dL)] [13]. Despite being widely used, it is not clear whether this formula needs adjustments for specific populations, such as critically ill patients [13]. For these patients, ionized calcium should be tested, if the test is available [13].
The prevalence of hypercalcemia in the emergency department is 0.6% and overall prevalence in the general population is 1 in 1000 individuals [13]. Most cases are caused by primary hyperparathyroidism or malignancies, corresponding to 80–90% of all cases [13–15]. Although hypercalcemia is not as prevalent as other electrolyte disorders, it causes high morbidity. Approximately 20% of cancer patients will develop hypercalcemia, which is more common in advanced disease [16]. In addition to its high prevalence, hypercalcemia is also related to increased mortality in people with cancer [11,17,18]. Primary hyperparathyroidism is more frequent in women than men, with a prevalence of 0.86% per year in the general population [19].
Since hypercalcemia diagnosis is based on laboratory findings and most patients are asymptomatic, it is important to identify people at risk of developing hypercalcemia. Calcium levels should be assessed in patients with personal histories of nephrolithiasis, early osteopenia or osteoporosis, or use of medications associated with hypercalcemia such as calcium, vitamin D and A supplements, thiazide diuretics, and lithium carbonate [20]. Serum calcium should also be evaluated in patients with bone, kidney, or intestine diseases. However, most calcium disturbances are still diagnosed during routine laboratory exams [19,20].
Etiology
The next step after diagnosing hypercalcemia is to determine its cause; correct diagnosis allows effective and often definitive treatment. Table 1 summarizes the causes of hypercalcemia [2,14]. The main causes are primary hyperparathyroidism and malignancy-associated hypercalcemia [2,14]. To minimize health expenses and systematize diagnostic evaluation, we should start by investigating these 2 differential diagnoses.
High calcium concentration generally suggests malignancy-associated hypercalcemia, whereas slightly or moderately increased serum calcium suggests hyperparathyroidism [21]. However, additional investigation is always indicated [21]. Neoplasia is the most frequent cause of hypercalcemia in hospitalized and emergency room patients and primary hyperparathyroidism is responsible for most cases outside the hospital setting [14,20,22].
Primary hyperparathyroidism leads to excessive PTH production. In approximately 80% of cases, hyperparathyroidism is caused by benign hyperplasia (adenoma) of 1 parathyroid gland. In 20% of cases, overproduction occurs in 2 or 3 glands, and in a small proportion of patients, hyperplasia occurs in all 4 glands. Less than 1% of cases are caused by parathyroid cancer, with a similar prevalence in men and women; patients are usually younger, between 40 and 50 years old [12,19,22]. In these cases, serum calcium concentration is usually higher than in benign hyperplasia. The prevalence of primary hyperparathyroidism in the United States is higher in women (23/10 000 individuals) than men (8.5/10 000 individuals) [19,23]. The peak of disease occurs in the seventh decade of life [24]. Risk factors for primary hyperparathyroidism include radiation to the head and neck in childhood and prolonged lithium use. Although most cases of primary hyperparathyroidism are sporadic, 5% to 10% of cases are related to hereditary syndromes such as multiple endocrine neoplasia, hyperparathyroidism jaw tumor syndrome, familial isolated hyperparathyroidism, neonatal severe primary hyperparathyroidism, and non-syndromic primary hyperparathyroidism [11,13]
The second most frequent etiology is malignancy-associated hypercalcemia. It occurs in 10% to 30% of cancer patients. Hypercalcemia is an important prognostic marker [25], with approximately 50% mortality at 1 month and up to 75% at 3 months after diagnosis. Cancer patient quality of life is also impacted by calcium levels [26]. The mechanisms of hypercalcemia, in order of frequency, are the production of PTH-related protein (PTHrp), bone metastasis, excessive activation of extrarenal vitamin D, and ectopic PTH secretion [17,25].
PTHrp is structurally similar to PTH; it binds to the same PTH bone receptors stimulating the synthesis of receptor activator of nuclear factor-kappa B ligand (RANKL) and activating osteoclasts, thus releasing calcium into the circulation [11]. Additionally, PTHrp increases renal tubular reabsorption of calcium. Differently from PTH, PTHrp does not increase renal vitamin D hydroxylation or calcium absorption from the intestine. Cancer sites most related to PTHrp production are the head, neck, esophagus, lung, kidney, breast, bladder, endometrium, ovaries, and intestines [11].
The second most frequent cause of malignancy-associated hypercalcemia is bone metastases, which represents approximately 20% of cases. Bone destruction by tumors or metastasis was previously believed to be the primary mechanism, but it is currently known that bone destruction is a secondary mechanism. Metastases produce several inflammatory cytokines, such as interleukin (IL)-1, IL-3, IL-6, tumor necrosis factor alpha, and transforming growth factor beta, that stimulate osteoblasts to produce RANKL and activate osteoclasts to release calcium into the circulation [18,11]. Additionally, some types of metastases, such as breast cancer metastases, can induce local PTHrp production. The cancers most frequently related to this type of mechanism are Hodgkin and non-Hodgkin lymphoma, breast cancer, and multiple myeloma [18]. Less frequently, ectopic PTH production can occur in lung and ovarian cancers, and some types of tumors such as lymphomas and ovarian cancer can cause extrarenal vitamin D activation [18].
The third most frequent hypercalcemia etiology is related to vitamin D status. Despite being rarely related to hypercalcemia in the past, it has become more frequent as vitamin D supplementation is increasingly common in clinical practice [14]. Indiscriminate vitamin D supplementation can increase intestinal calcium absorption, leading to hypervitaminosis and thus hypercalcemia [14].
Additionally, hypocalcemia or chronic kidney disease stimulates the parathyroid to produce PTH, which is called secondary hyperparathyroidism. When this stimulation is intense or prolonged, the glands start autonomous PTH production, no longer depending on exogenous stimulation; this is called tertiary hyperparathyroidism [27].
Granulomatous diseases such as tuberculosis, Hansen disease, cryptococcosis, histoplasmosis, sarcoidosis, paracoccidioidomycosis, and granulomatosis with polyangiitis can also cause hypercalcemia by producing 1,25[OH]2D. Macrophages in the granulomas produce 1-α-hydroxylase, which converts 25[OH]D into its active form, increasing intestinal calcium absorption [28].
Exogenous vitamin D intoxication and extrarenal vitamin D production can lead to PTH suppression and increase 1,25[OH]2D. Serum 25[OH]D level is normal in extrarenal vitamin D production, whereas the excessive vitamin D ingestion leads to high levels. In fact, there is no other cause for hypercalcemia associated with vitamin D levels above upper limits than exogenous vitamin D intoxication [22,29].
Although less frequent, some medications, such as lithium, thiazide diuretics, aminophylline, tamoxifen, and vitamins A and D may be related to hypercalcemia [22]. Endocrinological diseases, such as pheochromocytoma, adrenal insufficiency and, more relevantly, hyperthyroidism, can also be associated with hypercalcemia. In Graves disease, increased activation of osteoclasts by triiodothyronine (T3) can lead to hypercalcemia in up to 20% of patients. In general, thyrotoxicosis-induced hypercalcemia is mild and asymptomatic [22,30–32].
Although rare, familial hypocalciuric hypercalcemia also presents hypercalcemia. It is characterized by a mutation in the calcium-sensing receptor gene which increases the set point for calcium sensing, making the parathyroid less sensitive to calcium, so a higher serum calcium level is required to induce PTH release [33].
Clinical Manifestation
The signs and symptoms of hypercalcemia are related to calcium levels, with higher values leading to more pronounced symptoms. Onset time is also important in the development of symptoms; acute onset is frequently associated with more symptoms than chronic onset, which allows the body to adapt to the new metabolic condition [17]. Usually, calcium values between 10.5 and 12 mg/dL rarely cause symptoms, values greater than 14 mg/dL are associated with more frequent and severe symptoms, and calcium values higher than 15 mg/dL increase the risk of cardiac arrest [21].
As mentioned previously, most patients are asymptomatic and diagnosed during routine tests. Only a small percentage of hypercalcemia patients present with exuberant and severe symptoms, which involve neurological, muscular, gastrointestinal, renal, and cardiovascular systems, as seen in Table 2 [1,2,13,34,35].
At initial assessment, hypercalcemia is usually marked by dehydration, which occurs through various mechanisms. Hypercalcemia causes renal afferent arteriole vasoconstriction, which decreases renal blood flow and glomerular filtration rate (GFR), further reducing the ability of the kidneys to excrete the excess calcium [36–39]. Also, hypercalcemia affects tubular function by inhibiting the sodium-chloride transporter and increasing sodium and chloride loss, thus leading to volume depletion [37]. Additionally, hypercalcemia downregulates aquaporin 2 water channels in the collecting ducts, which contributes to polyuria [36]. The volume contraction causes additional renal sodium retention by activating the renin-angiotensin-aldosterone system, and leads to reduced urinary calcium excretion, by stimulating its reabsorption in the proximal renal tubule, since the transport of these 2 ions is linked [39].
Chronic hypercalcemia presents with less exuberant symptoms. Nephrolithiasis, osteopenia, and osteoporosis are common clinical findings. Patients with primary hyperparathyroidism have lower bone mineral density than individuals of the same sex and age without hyperparathyroidism. Approximately 15% of hyperparathyroidism patients have osteopenia in the lumbar spine. Depression and memory loss are also associated with chronic hypercalcemia, but the causal link between these conditions and parathyroid disease is uncertain [19].
Complementary Tests and Diagnosis
Initial evaluation of hypercalcemia patients involves assessing serum levels of PTH, vitamin D, total proteins and fractions, phosphorus and creatinine, and electrocardiography. Measurement of 25[OH]D can differentiate between excess vitamin D intake and extrarenal vitamin D activation [22,29]. From these results, it may be necessary to perform other laboratory tests, such as PTHrp and urinary calcium. Although most cases of malignancy-associated hypercalcemia are related to PTHrp production, it is rarely necessary to assess PTHrp to establish a diagnosis. Furthermore, PTHrp evaluation is expensive, difficult to perform, and not widely available [22,29].
Urinary calcium and urinary creatinine levels can be helpful in calculating calcium excretion by the formula UrCa x SerCr/SerCa x UrCr, where UrCa is urinary calcium (mg/24 h), SerCa is serum calcium (mg/dL), UrCr is urinary creatinine (mg/24 h), and SerCr is serum creatinine (mg/dL); this is useful in differentiating primary hyperparathyroidism from familial hypocalciuric hypercalcemia, its main differential diagnosis. A result <0.01 suggests familial hypocalciuric hypercalcemia and a result of >0.01 suggests primary hyperparathyroidism [13].
It is important to highlight that imaging exams are not essential for diagnosing primary hyperparathyroidism, but they could be performed to locate the affected gland and for a surgical planning. Additionally, imaging exams may be performed to diagnose nephrolithiasis and densitometry is mandatory for evaluating bone mineral density [13].
The steps for etiological investigation of hypercalcemia are summarized in Figure 2.
Treatment
Treatment of hypercalcemia involves intravenous hydration, drugs such as bisphosphonates and denosumab to reduce bone resorption, corticosteroids, and, most importantly, treatment of the underlying cause [39–42].
Hydration is the first measure in hypercalcemia treatment. The fluid therapy should replace sodium and water deficits and stimulate a sodium-linked calcium diuresis in the proximal renal tubule, as these 2 ions move in parallel at this part of the nephron [39–41]. Heller et al [40] showed that saline alone reduces calcium by an average of 2.4 mg/dL; this may be enough to correct hypercalcemia in mild cases. In this trial, all patients displayed clinical improvement after venous hydration [39,40].
There are no randomized clinical trials comparing different types of fluid in hypercalcemic patients; considering the mechanisms involved in volume depletion and the sodium-calcium transport relationship in proximal tubules, we should prioritize 0.9% saline, because it contains more sodium than balanced solutions. However, it is important to emphasize that the type of fluid should always be individualized according to electrolytes and the presence of acid-base disorders [42].
We usually administer 0.9% saline intravenously, starting with 1 to 2 L, followed by an infusion of 200 to 300 mL/h. The hydration should ensure a diuresis of 100 to 150 mL/h, a urinary flow that promotes effective calciuresis [17,28]. If the diuresis is unsatisfactory, hydration should be adjusted, always aiming at the above-mentioned diuresis.
In the past, furosemide was a standard of care for hypercalcemia treatment. In a review, LeGrand et al [43] found no studies since 1983 investigating furosemide dose or efficiency in hypercalcemia management. The effect of furosemide in calcium diuresis is limited and may aggravate dehydration and electrolytic disturbances; its use should therefore be limited to patients with volume overload [18].
The second cornerstone of hypercalcemia treatment is bisphosphonates. Saline with bisphosphonates is the first-line treatment, with level-one evidence for hypercalcemia [43]. Most trials with bisphosphonates have only included patients with calcium levels above 12 mg/dL; for this reason, hydration only is initially recommended in mild hypercalcemia. In moderate or severe hypercalcemia, or in very symptomatic patients, bisphosphonates should be added to intravenous hydration. The 2 options approved for hypercalcemia treatment are pamidronate and zoledronic acid. These medications have a high affinity for hydroxyapatite and are quickly deposited in the bone after administration. Bisphosphonates are internalized by osteoclasts, blocking structural proteins and inducing apoptosis of these cells. As a result, bone reabsorption is decreased, and calcium levels are consequently reduced [11,28].
Pamidronate was approved by the Food and Drug Administration (FDA) for clinical use in 1991. In a systematic review of bisphosphonate use in malignancy-related hypercalcemia, Saunders et al [44] showed that bisphosphonates are highly effective, with over 70% of patients reaching normocalcemia. Small studies showed no differences between 30, 60, or 90 mg pamidronate [45,46], but a large, double-blind study suggested increased efficacy at higher doses [47]. Although several authors recommend lower doses for moderate hypercalcemia, evidence suggests that higher bisphosphonate doses should be considered, regardless of initial calcium value [44].
Zoledronic acid is more potent than pamidronate in normalizing calcium levels. Major et al [48] conducted 2 concurrent randomized controlled trials that compared 4 mg and 8 mg of zoledronic acid with 90 mg of pamidronate; both zoledronic acid doses were more effective than pamidronate, with 88.4%, 86.7% and 69.7% of patients achieving normal serum calcium concentration after 10 days, respectively [48]. However, unlike zoledronic acid, pamidronate can be used in association with calcitonin, and promotes a faster response [11,25,49].
The usual pamidronate dose is between 60 and 90 mg intravenously, and infusion should be given in 2 to 4 h. In patients with GFR <30 mL/min, extended infusion time and reduced dose should be considered [34], but there are not enough clinical trials to support this recommendation. The average time to reach normocalcemia is 4 days and the effect of the medication lasts around 18 days. The dose of zoledronic acid is 4 to 8 mg intravenously, infused over 15 min. In patients with GRF between 30 and 60 mL/min, the dose should be reduced; zoledronic acid is not recommended for patients with a GFR lower than 30 mL/min [34]. The effect on calcemia can be seen in less than 3 days and it lasts approximately 32 days [11,28,50].
Calcitonin has been approved by the FDA for treating osteoporosis since 1975 and has more recently become part of the therapeutic arsenal for hypercalcemia when a rapid decrease in calcium levels is needed. The action mechanism consists of decreasing osteoclast activity and renal calcium reabsorption, reducing calcium levels. In some countries, calcitonin is not widely available. It has the advantage of a faster effect (12 to 24 h), but it downregulates calcitonin receptors in the bone and kidneys, inducing tachyphylaxis in 48 h [34]; therefore, its use in combination with pamidronate is controversial [2]. It is administered intramuscularly or subcutaneously at a dose of 4 U/kg every 12 h, or even every 6 h if the response is not satisfactory. The effect of isolated calcitonin in hypercalcemia treatment has been poorly studied [11,50].
The use of corticosteroids is limited to the treatment of some cases of malignancy-related hypercalcemia, especially in multiple myeloma, lymphomas, and breast cancer. Corticosteroids decrease the production of inflammatory mediators by the bones and therefore decrease osteoclast activity. They can also be used in vitamin D-mediated hypercalcemia because they reduce 1-α-hydroxylase activity. The recommended dose is 200 to 300 mg hydrocortisone daily or 40 to 60 mg prednisone per day for 3 to 5 days. It should be associated with another therapeutic strategy and interrupted if there is an unsatisfactory response in 10 days [11,50]
Denosumab was approved by the FDA in 2010, initially for treating osteoporosis and skeletal-related events in patients with bone metastases; only in 2018 was denosumab approved for malignancy-related hypercalcemia that is unresponsive to bisphosphonates. Denosumab is a monoclonal antibody against RANKL. Blockage of RANKL prevents maturation and the function and survival of osteoclasts and therefore decreases bone resorption [51]. There have been some case reports since 2012 on the effectiveness of denosumab in hypercalcemia-related malignancy [52–56]. In a randomized, double-blind, clinical trial comparing denosumab and zoledronic acid in patients with advanced cancer and bone metastases or with multiple myeloma, Diel et al [57] showed that denosumab is more effective than zoledronic acid in delaying and preventing hypercalcemia [57]. The recommended dose is a subcutaneous 120 mg denosumab weekly for the first month and then once every 4 weeks [58–63].
Although not frequently reported, hemodialysis may be indicated in some cases [64]. It is an alternative for patients refractory to standard management, or with severe kidney injury or congestive heart failure, in whom hydration cannot be safely performed, or if urgent reduction in calcium is needed, such as in patients with severe neurological symptoms [18,65,66].
Final Considerations
Hypercalcemia is an important electrolyte disturbance due to its high associated morbidity and mortality, and the potentially severe consequences. Hypercalcemia can be diagnosed in different medical scenarios, such as in primary care or the emergency room. As most patients are asymptomatic, a high suspicion for the disorder is essential for an early and correct diagnosis. The etiology of hypercalcemia should always be determined to improve prognosis and the use of financial resources. Although treatment is mainly based on intravenous hydration and bisphosphonates administration, it should be individualized in specific situations. Treatment for patients with very high serum calcium or clinically relevant symptoms must be started as soon as possible, even before any diagnostic investigation.
Conclusions
As hypercalcemia is a potentially life-threatening condition and patients might not have any symptoms, we should keep a high suspicion for the diagnosis, mainly in emergency room scenarios. The etiological diagnosis should always be sought; however, starting general treatment quickly is essential for the prognosis of patients.
Figures
![Calcium Homeostasis. Vitamin D is produced by the skin after exposure to ultraviolet B radiation and a small amount comes from diet. In the skin, vitamin D2 and D3 undergo hydroxylation in the liver by 25-hydroxylase, generating 25-hydroxyvitamin D (25[OH]D). In the kidneys, 1-α-hydroxylase converts 25[OH]D into 1,25-dihydroxyvitamin D (1,25[OH]2D). This form of vitamin D increases intestinal calcium absorption. Parathyroid hormone (PTH) is produced by the parathyroid glands. Decreased serum calcium concentration stimulates PTH release, which increases calcium bone absorption, renal calcium reabsorption, renal hydroxylation of 25[OH]D to 1,25[OH]2D, and, less importantly, intestinal calcium absorption. Created by the authors with Power Point, version 2013, manufactured by Microsoft.](https://jours.isi-science.com/imageXml.php?i=medscimonit-28-e935821-g001.jpg&idArt=935821&w=1000) Figure 1. Calcium Homeostasis. Vitamin D is produced by the skin after exposure to ultraviolet B radiation and a small amount comes from diet. In the skin, vitamin D2 and D3 undergo hydroxylation in the liver by 25-hydroxylase, generating 25-hydroxyvitamin D (25[OH]D). In the kidneys, 1-α-hydroxylase converts 25[OH]D into 1,25-dihydroxyvitamin D (1,25[OH]2D). This form of vitamin D increases intestinal calcium absorption. Parathyroid hormone (PTH) is produced by the parathyroid glands. Decreased serum calcium concentration stimulates PTH release, which increases calcium bone absorption, renal calcium reabsorption, renal hydroxylation of 25[OH]D to 1,25[OH]2D, and, less importantly, intestinal calcium absorption. Created by the authors with Power Point, version 2013, manufactured by Microsoft.
Figure 1. Calcium Homeostasis. Vitamin D is produced by the skin after exposure to ultraviolet B radiation and a small amount comes from diet. In the skin, vitamin D2 and D3 undergo hydroxylation in the liver by 25-hydroxylase, generating 25-hydroxyvitamin D (25[OH]D). In the kidneys, 1-α-hydroxylase converts 25[OH]D into 1,25-dihydroxyvitamin D (1,25[OH]2D). This form of vitamin D increases intestinal calcium absorption. Parathyroid hormone (PTH) is produced by the parathyroid glands. Decreased serum calcium concentration stimulates PTH release, which increases calcium bone absorption, renal calcium reabsorption, renal hydroxylation of 25[OH]D to 1,25[OH]2D, and, less importantly, intestinal calcium absorption. Created by the authors with Power Point, version 2013, manufactured by Microsoft. ![Etiological Investigation of Hypercalcemia. UrCa: urinary calcium (mg/24 h); SerCa: serum calcium (mg/dL); UrCr: urinary creatinine (mg/24 h); SerCr: serum creatinine (mg/dL); PTH: parathyroid hormone; 25[OH]D: 25-hydroxyvitamin D; 1,25[OH]2D: 1,25-dihydroxyvitamin D. Created by the authors with Power Point, version 2013, manufactured by Microsoft.](https://jours.isi-science.com/imageXml.php?i=medscimonit-28-e935821-g002.jpg&idArt=935821&w=1000) Figure 2. Etiological Investigation of Hypercalcemia. UrCa: urinary calcium (mg/24 h); SerCa: serum calcium (mg/dL); UrCr: urinary creatinine (mg/24 h); SerCr: serum creatinine (mg/dL); PTH: parathyroid hormone; 25[OH]D: 25-hydroxyvitamin D; 1,25[OH]2D: 1,25-dihydroxyvitamin D. Created by the authors with Power Point, version 2013, manufactured by Microsoft.
Figure 2. Etiological Investigation of Hypercalcemia. UrCa: urinary calcium (mg/24 h); SerCa: serum calcium (mg/dL); UrCr: urinary creatinine (mg/24 h); SerCr: serum creatinine (mg/dL); PTH: parathyroid hormone; 25[OH]D: 25-hydroxyvitamin D; 1,25[OH]2D: 1,25-dihydroxyvitamin D. Created by the authors with Power Point, version 2013, manufactured by Microsoft. 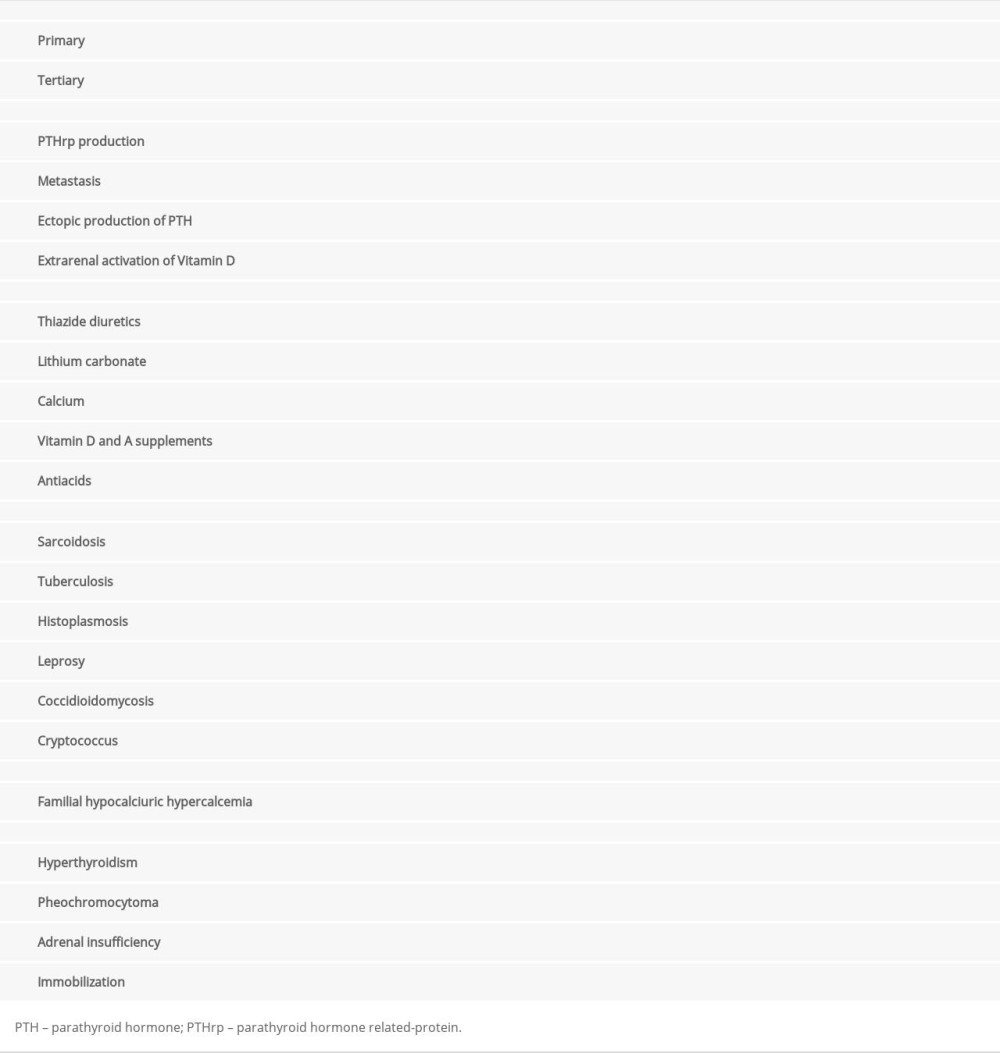

References
1. Basso SMM, Lumachi F, Nascimben F, Treatment of acute hypercalcemia: Med Chem, 2012; 8(4); 564-68
2. Chang WTW, Radin B, McCurdy MT, Calcium, magnesium, and phosphate abnormalities in the emergency department: Emerg Med Clin North Am, 2014; 32(2); 349-66
3. Endo M, Calcium ion as a second messenger with special reference to excitation-contraction coupling: J Pharmacol Sci, 2006; 100(5); 519-24
4. Singh S, Dodt J, Volkers P, Structure functional insights into calcium binding during the activation of coagulation factor XIII A: Sci Rep, 2019; 9(1); 11324
5. Kuo IY, Ehrlich BE, Signaling in muscle contraction: Cold Spring Harb Perspect Biol, 2015; 7(2); a006023
6. Webb RC, Bohr DF, Mechanism of membrane stabilization by calcium in vascular smooth muscle: Am J Physiol-Cell Physiol, 1978; 235(5); C227-32
7. Ariyan CE, Sosa JA, Assessment and management of patients with abnormal calcium: Crit Care Med, 2004; 32; S146-54
8. Žofková I, Hypercalcemia. Pathophysiological aspects: Physiol Res, 2016; 65(1); 1-10
9. Kinder BK, Stewart AF, Hypercalcemia: Curr Probl Surg, 2002; 39(4); 360-447
10. Bollerslev J, Pretorius M, Heck A, Parathyroid hormone independent hypercalcemia in adults: Best Pract Res Clin Endocrinol Metab, 2018; 32(5); 621-38
11. Goldner W, Cancer-related hypercalcemia: J Oncol Pract, 2016; 12(5); 426-32
12. Bilezikian JP, Bandeira L, Khan A, Cusano NE, Hyperparathyroidism: Lancet Lond Engl, 2018; 391(10116); 168-78
13. Turner JJO, Hypercalcaemia – presentation and management: Clin Med, 2017; 17(3); 270-73
14. Banu S, Batool S, Sattar S, Masood MQ, Malignant and non-malignant causes of hypercalcemia: A retrospective study at a tertiary care hospital in Pakistan: Cureus, 2021; 13(6); e15845
15. Motlaghzadeh Y, Bilezikian JP, Sellmeyer DE, Rare causes of hypercalcemia: 2021 update: J Clin Endocrinol Metab, 2021; 106(11); 3113-28
16. Spierling A, Kikano EG, Chagarlamudi K, Imaging features of hypercalcemia: A primer for emergency radiologists: Clin Imaging, 2021; 80; 215-24
17. Klemencic S, Perkins J, Diagnosis and management of oncologic emergencies: West J Emerg Med, 2019; 20(2); 316-22
18. McCurdy MT, Shanholtz CB, Oncologic emergencies: Crit Care Med, 2012; 40(7); 2212-22
19. Insogna KL, Primary hyperparathyroidism: N Engl J Med, 2018; 379(11); 1050-59
20. Jacobi J, Management of endocrine emergencies in the ICU: J Pharm Pract, 2019; 32(3); 314-326
21. Acharya R, Winters DM, Rowe C, An unusual case of severe hypercalcemia: As dehydrated as a bone: J Community Hosp Intern Med Perspect, 2021; 11(1); 135-38
22. Endres DB, Investigation of hypercalcemia: Clin Biochem, 2012; 45(12); 954-63
23. Kim RC, Roch AM, Birdas TJ, Hypercalcemic crisis caused by a parathyroid mass requiring thoracoscopic resection: AACE Clin Case Rep, 2021; 7(4); 264-67
24. Eufrazino C, Veras A, Bandeira F, Epidemiology of primary hyperparathyroidism and its non-classical manifestations in the city of Recife, Brazil: Clin Med Insights Endocrinol Diabetes, 2013; 6; CMED.S13147
25. Gonciulea AR, Wang Y, Bikle DD, Sellmeyer DE, Hypercalcemia in non-Hodgkin’s lymphoma due to cosecretion of PTHrP and 1,25-dihydroxyvitamin D: Osteoporos Int, 2021; 32(12); 2587-92
26. O’Callaghan S, Yau H, Treatment of malignancy-associated hypercalcemia with cinacalcet: A paradigm shift: Endocr Connect, 2021; 10(1); R13-24
27. Palumbo V, Damiano G, Messina M, Tertiary hyperparathyroidism: A review: Clin Ter, 2021(3); 241-46
28. Muller I, Premawardhana LD, Hypercalcaemia with undetectable parathormone levels: BMJ, 2018; 363; k4074
29. Marcinowska-Suchowierska E, Kupisz-Urbańska M, Łukaszkiewicz J, Vitamin D toxicity – a clinical perspective: Front Endocrinol, 2018; 9; 550
30. Baxter JD, Hypercalcemia of thyrotoxicosis: Ann Intern Med, 1966; 65(3); 429
31. Chen K, Xie Y, Zhao L, Mo Z, Hyperthyroidism-associated hypercalcemic crisis: A case report and review of the literature: Medicine (Baltimore), 2017; 96(4); e6017
32. Schneider AB, Sherwood LM, Calcium homeostasis and the pathogenesis and management of hypercalcemic disorders: Metabolism, 1974; 23(10); 975-1007
33. Afzal M, Kathuria P, Familial hypocalciuric hypercalcemia: StatPearls, 2021 Available from: http://www.ncbi.nlm.nih.gov/books/NBK459190/
34. Ahmad S, Kuraganti G, Steenkamp D, Hypercalcemic crisis: A clinical review: Am J Med, 2015; 128(3); 239-45
35. Thillainadesan S, Twigg SM, Perera N, Prevalence, causes and associated mortality of hypercalcaemia in modern hospital care: Intern Med J, 2021; imj.15402 [Online ahead of print]
36. Pawar NH, Chiam PPS, Tan JHY, Acute kidney injury, hypercalcemia, and osteolytic lesions: A familiar triad with a rare cause complicated by posterior reversible encephalopathy syndrome: Am J Kidney Dis, 2017; 70(5); A12-15
37. Benabe JE, Hypercalcemic nephropathy: Arch Intern Med, 1978; 138(5); 777
38. Stewart AF, Hypercalcemia associated with cancer: N Engl J Med, 2005; 352(4); 373-79
39. Ralston S, Medical management of hypercalcaemia: Br J Clin Pharmacol, 1992; 34(1); 11-20
40. Heller SR, Hosking DJ, Renal handling of calcium and sodium in metastatic and non-metastatic malignancy: BMJ, 1986; 292(6520); 583-86
41. Meyer WJ, Transbol I, Bartter FC, Delea C, Control of calcium absorption: Effect of sodium chloride loading and depletion: Metabolism, 1976; 25(9); 989-93
42. Hoorn EJ, Intravenous fluids: balancing solutions: J Nephrol, 2017; 30(4); 485-92
43. LeGrand SB, Leskuski D, Zama I, Narrative review: Furosemide for hypercalcemia: An unproven yet common practice: Ann Intern Med, 2008; 149(4); 259
44. Saunders Y, Ross JR, Broadley KE, Systematic review of bisphosphonates for hypercalcaemia of malignancy: Palliat Med, 2004; 18(5); 418-31
45. Gallacher SJ, Ralston SH, Fraser WD, A comparison of low versus high dose pamidronate in cancer-associated hypercalcaemia: Bone Miner, 1991; 15(3); 249-56
46. Body JJ, Magritte A, Seraj F, Aminohydroxypropylidene bisphosphonate (APD) treatment for tumor-associated hypercalcemia: A randomized comparison between a 3-day treatment and single 24-hour infusions: J Bone Miner Res, 2009; 4(6); 923-28
47. Nussbaum SR, Younger J, Vandepol CJ, Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: Comparison of 30-, 60-, and 90-mg dosages: Am J Med, 1993; 95(3); 297-304
48. Major P, Lortholary A, Hon J, Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: A pooled analysis of two randomized, controlled clinical trials: J Clin Oncol, 2001; 19(2); 558-67
49. Minisola S, Pepe J, Piemonte S, Cipriani C, The diagnosis and management of hypercalcaemia: BMJ, 2015; 350; h2723
50. Clines GA, Mechanisms and treatment of hypercalcemia of malignancy: Curr Opin Endocrinol Diabetes Obes, 2011; 18(6); 339-46
51. Thosani S, Hu MI, Denosumab: A new agent in the management of hypercalcemia of malignancy: Future Oncol, 2015; 11(21); 2865-71
52. Bech A, de Boer H, Denosumab for tumor-Induced hypercalcemia complicated by renal failure: Ann Intern Med, 2012; 156(12); 906
53. Boikos SA, Hammers HJ, Denosumab for the treatment of bisphosphonate-refractory hypercalcemia: J Clin Oncol, 2012; 30(29); e299
54. Adhikaree J, Newby Y, Sundar S, Denosumab should be the treatment of choice for bisphosphonate refractory hypercalcaemia of malignancy: Case Rep, 2014; 2014 bcr2013202861
55. Freeman A, El-Amm J, Aragon-Ching JB, Use of denosumab for renal cell carcinoma-associated malignant hypercalcemia: A case report and review of the literature: Clin Genitourin Cancer, 2013; 11(4); e24-26
56. Dietzek A, Connelly K, Cotugno M, Denosumab in hypercalcemia of malignancy: A case series: J Oncol Pharm Pract, 2015; 21(2); 143-47
57. Diel IJ, Body JJ, Stopeck AT, The role of denosumab in the prevention of hypercalcaemia of malignancy in cancer patients with metastatic bone disease: Eur J Cancer, 2015; 51(11); 1467-75
58. Polyzos SA, Makras P, Tournis S, Anastasilakis AD, Off-label uses of denosumab in metabolic bone diseases: Bone, 2019; 129; 115048
59. Feldenzer KL, Sarno J, Hypercalcemia of malignancy: J Adv Pract Oncol, 2018; 8(5); 496-504
60. Mirrakhimov A, Hypercalcemia of malignancy: An update on pathogenesis and management: North Am J Med Sci, 2015; 7(11); 483
61. Glezerman IG, Sternlicht H, Hypercalcemia of malignancy and new treatment options: Ther Clin Risk Manag, 2015; 11; 1779-88
62. Thosani S, Hu MI, Denosumab: A new agent in the management of hypercalcemia of malignancy: Future Oncol, 2015; 11(21); 2865-71
63. Fallah-Rad N, Morton AR, Managing hypercalcaemia and hypocalcaemia in cancer patients: Curr Opin Support Palliat Care, 2013; 7(3); 265-71
64. Loh HH, Mohd Noor N, The use of hemodialysis in refractory hypercalcemia secondary to parathyroid carcinoma: Case Rep Crit Care, 2014; 2014; 140906
65. Rosner MH, Dalkin AC, Onco-Nephrology: The pathophysiology and treatment of malignancy-associated hypercalcemia: Clin J Am Soc Nephrol, 2012; 7(10); 1722-29
66. Basok AB, Rogachev B, Haviv YS, Vorobiov M, Treatment of extreme hypercalcaemia: The role of haemodialysis: BMJ Case Rep, 2018; 2018 bcr2017223772
Figures
Figure 1. Calcium Homeostasis. Vitamin D is produced by the skin after exposure to ultraviolet B radiation and a small amount comes from diet. In the skin, vitamin D2 and D3 undergo hydroxylation in the liver by 25-hydroxylase, generating 25-hydroxyvitamin D (25[OH]D). In the kidneys, 1-α-hydroxylase converts 25[OH]D into 1,25-dihydroxyvitamin D (1,25[OH]2D). This form of vitamin D increases intestinal calcium absorption. Parathyroid hormone (PTH) is produced by the parathyroid glands. Decreased serum calcium concentration stimulates PTH release, which increases calcium bone absorption, renal calcium reabsorption, renal hydroxylation of 25[OH]D to 1,25[OH]2D, and, less importantly, intestinal calcium absorption. Created by the authors with Power Point, version 2013, manufactured by Microsoft.Figure 2. Etiological Investigation of Hypercalcemia. UrCa: urinary calcium (mg/24 h); SerCa: serum calcium (mg/dL); UrCr: urinary creatinine (mg/24 h); SerCr: serum creatinine (mg/dL); PTH: parathyroid hormone; 25[OH]D: 25-hydroxyvitamin D; 1,25[OH]2D: 1,25-dihydroxyvitamin D. Created by the authors with Power Point, version 2013, manufactured by Microsoft. In Press
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
08 Mar 2024 : Laboratory Research
Evaluation of Retentive Strength of 50 Endodontically-Treated Single-Rooted Mandibular Second Premolars Res...Med Sci Monit In Press; DOI: 10.12659/MSM.944110
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952